

A network-based approach for predicting missing pathway interactions
- PMID: 22916002
- PMCID: PMC3420932
- DOI: 10.1371/journal.pcbi.1002640
A network-based approach for predicting missing pathway interactions
Abstract
Embedded within large-scale protein interaction networks are signaling pathways that encode response cascades in the cell. Unfortunately, even for well-studied species like S. cerevisiae, only a fraction of all true protein interactions are known, which makes it difficult to reason about the exact flow of signals and the corresponding causal relations in the network. To help address this problem, we introduce a framework for predicting new interactions that aid connectivity between upstream proteins (sources) and downstream transcription factors (targets) of a particular pathway. Our algorithms attempt to globally minimize the distance between sources and targets by finding a small set of shortcut edges to add to the network. Unlike existing algorithms for predicting general protein interactions, by focusing on proteins involved in specific responses our approach homes-in on pathway-consistent interactions. We applied our method to extend pathways in osmotic stress response in yeast and identified several missing interactions, some of which are supported by published reports. We also performed experiments that support a novel interaction not previously reported. Our framework is general and may be applicable to edge prediction problems in other domains.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
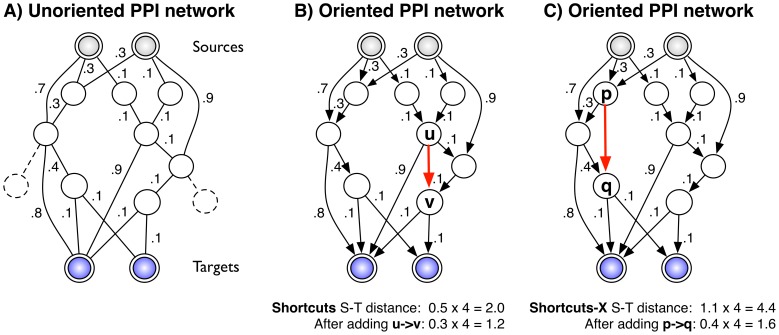
 hops from any source-target pair are purged (shown dashed in A). The red arrow indicates an edge prediction (
hops from any source-target pair are purged (shown dashed in A). The red arrow indicates an edge prediction ( ) that globally minimizes the distance between each source and target using the S
) that globally minimizes the distance between each source and target using the S . Here, the total hop-restricted distance between each source and target is higher (4.4) and the optimal edge,
. Here, the total hop-restricted distance between each source and target is higher (4.4) and the optimal edge,  reduces the distance to 1.6.
reduces the distance to 1.6.
 shows the number of edges added, and the
shows the number of edges added, and the  shows the new objective function cost as a percent of the original cost. Each new edge was added with weight 0.0. For S
shows the new objective function cost as a percent of the original cost. Each new edge was added with weight 0.0. For S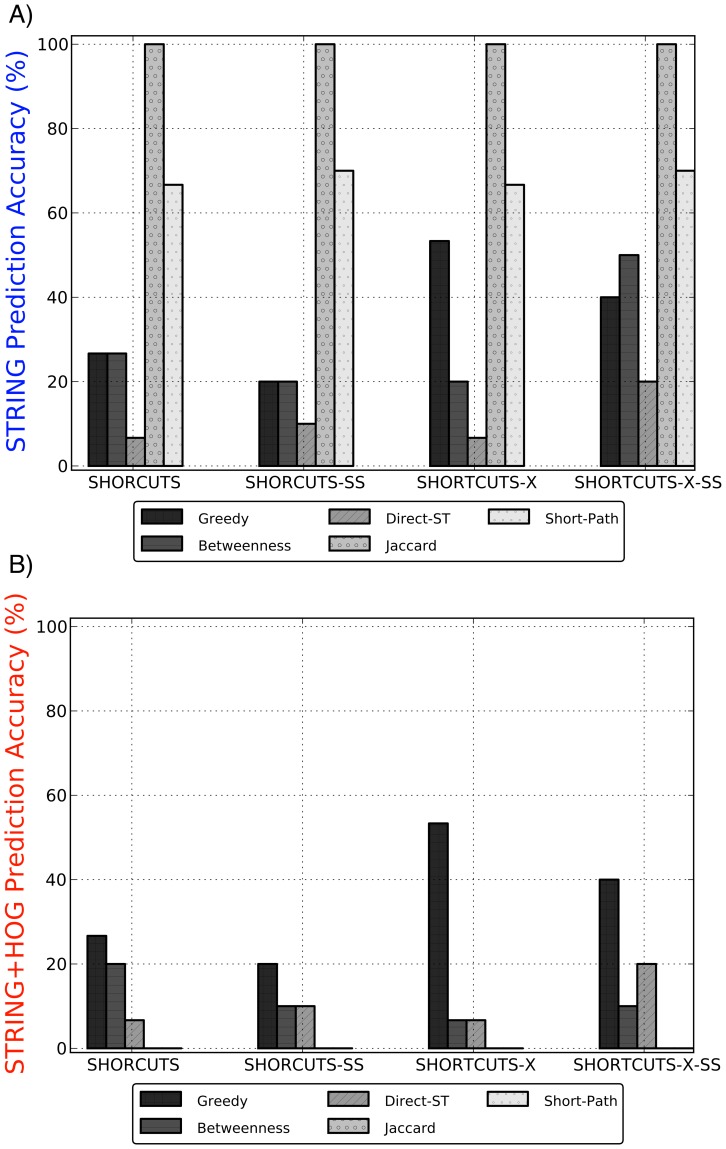
 shows the prediction accuracy, defined as the percentage of predictions (from amongst all
shows the prediction accuracy, defined as the percentage of predictions (from amongst all  million possible missing edges) that lied within the set of A) STRING potential edges, and B) STRING potential edges that also connected known HOG-related proteins. The global methods (Jaccard and Short-Path) make accurate predictions when not constrained to be HOG-relevant. The Greedy algorithm outperforms all methods in making high quality predictions that connect HOG proteins.
million possible missing edges) that lied within the set of A) STRING potential edges, and B) STRING potential edges that also connected known HOG-related proteins. The global methods (Jaccard and Short-Path) make accurate predictions when not constrained to be HOG-relevant. The Greedy algorithm outperforms all methods in making high quality predictions that connect HOG proteins.References
-
- Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, et al. (2006) Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440: 637–643. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
