

Advances in quantitative trait analysis in yeast
- PMID: 22916041
- PMCID: PMC3420948
- DOI: 10.1371/journal.pgen.1002912
Advances in quantitative trait analysis in yeast
Abstract
Understanding the genetic mechanisms underlying complex traits is one of the next frontiers in biology. The budding yeast Saccharomyces cerevisiae has become an important model for elucidating the mechanisms that govern natural genetic and phenotypic variation. This success is partially due to its intrinsic biological features, such as the short sexual generation time, high meiotic recombination rate, and small genome size. Precise reverse genetics technologies allow the high throughput manipulation of genetic information with exquisite precision, offering the unique opportunity to experimentally measure the phenotypic effect of genetic variants. Population genomic and phenomic studies have revealed widespread variation between diverged populations, characteristic of man-made environments, as well as geographic clusters of wild strains along with naturally occurring recombinant strains (mosaics). Here, we review these recent studies and provide a perspective on how these previously unappreciated levels of variation can help to bridge our understanding of the genotype-phenotype gap, keeping budding yeast at the forefront of genetic studies. Not only are quantitative trait loci (QTL) being mapped with high resolution down to the nucleotide, for the first time QTLs of modest effect and complex interactions between these QTLs and between QTLs and the environment are being determined experimentally at unprecedented levels using next generation techniques of deep sequencing selected pools of individuals as well as multi-generational crosses.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
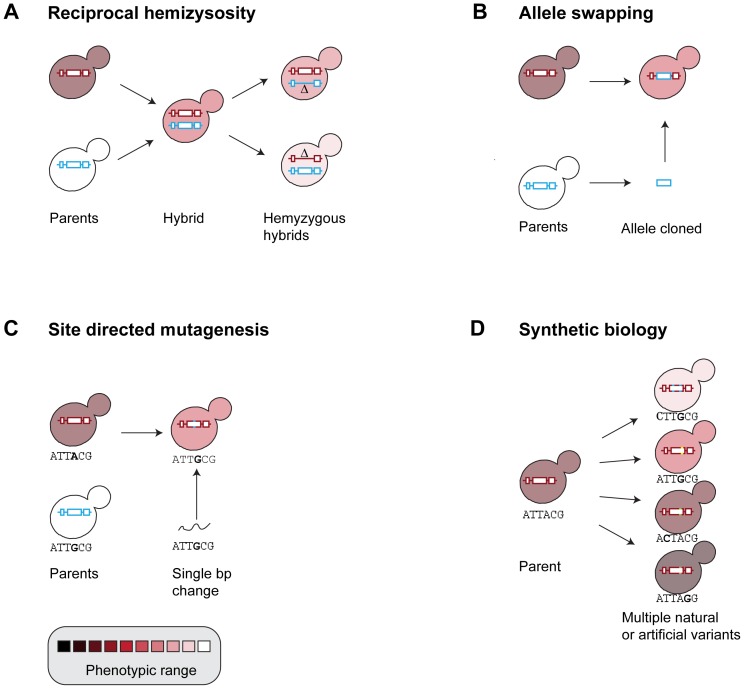
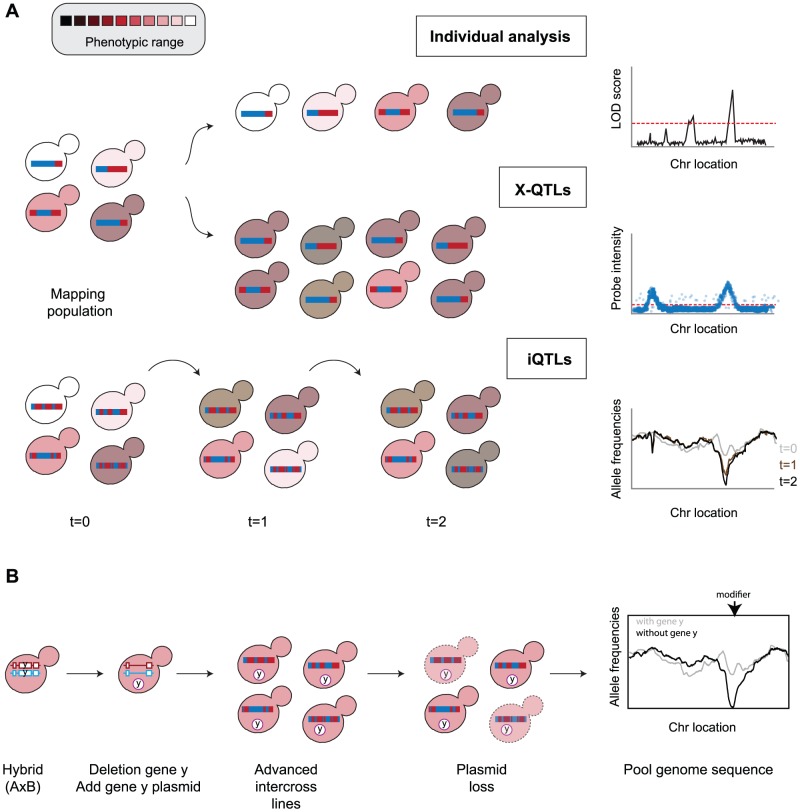
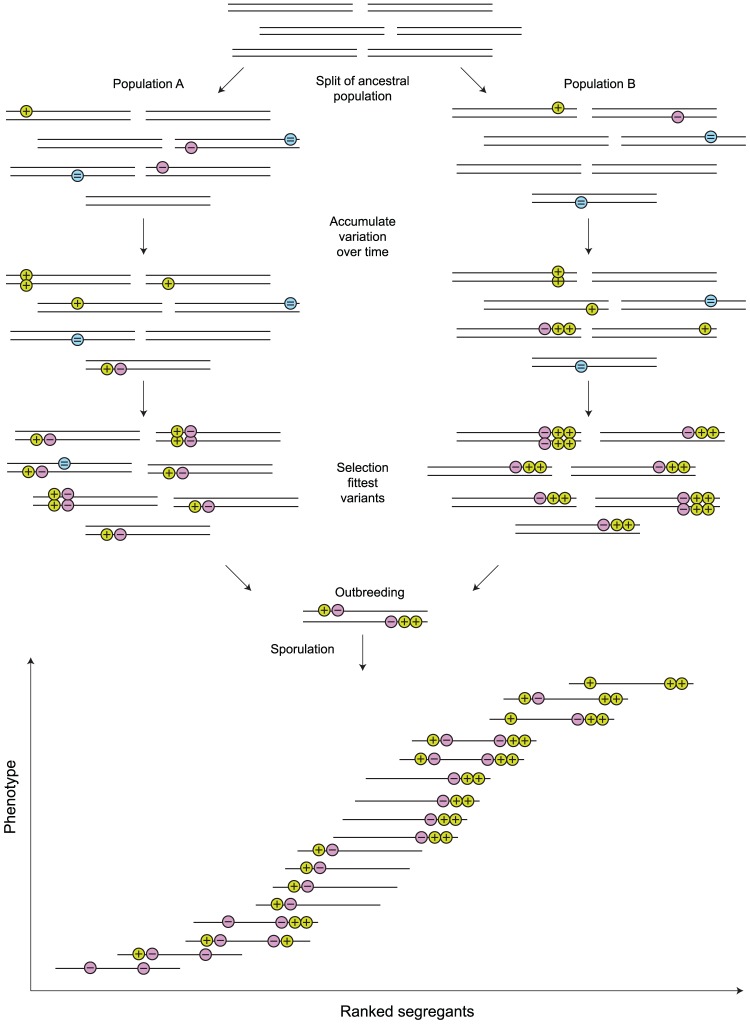
References
-
- Barnett JA (2007) A history of research on yeasts 10: foundations of yeast genetics. Yeast 24: 799–845. - PubMed
-
- Mackay TF, Stone EA, Ayroles JF (2009) The genetics of quantitative traits: challenges and prospects. Nat Rev Genet 10: 565–577. - PubMed
-
- Brem RB, Yvert G, Clinton R, Kruglyak L (2002) Genetic dissection of transcriptional regulation in budding yeast. Science 296: 752–755. - PubMed
-
- Steinmetz LM, Sinha H, Richards DR, Spiegelman JI, Oefner PJ, et al. (2002) Dissecting the architecture of a quantitative trait locus in yeast. Nature 416: 326–330. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
