

Critical role for mixed-lineage kinase 3 in acetaminophen-induced hepatotoxicity
- PMID: 22918968
- PMCID: PMC3477232
- DOI: 10.1124/mol.112.079863
Critical role for mixed-lineage kinase 3 in acetaminophen-induced hepatotoxicity
Abstract
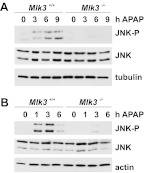
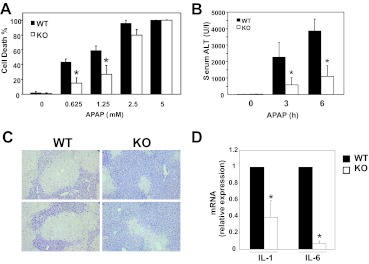
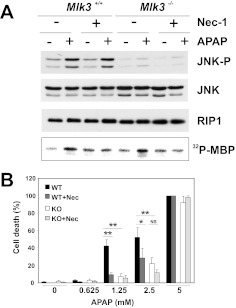
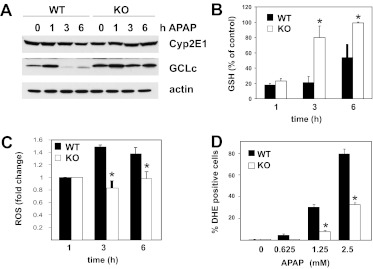
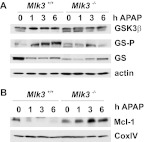
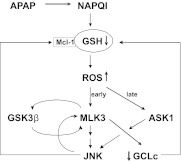
c-Jun NH(2)-terminal kinase (JNK) activation plays a major role in acetaminophen (APAP)-induced hepatotoxicity. However, the exact mechanism of APAP-induced JNK activation is incompletely understood. It has been established that apoptosis signal-regulating kinase 1 (ASK1) regulates the late phase of APAP-induced JNK activation, but the mitogen-activated protein kinase kinase kinase that mediates the initial phase of APAP-induced JNK activation has not been identified. Oxidative stress produced during APAP metabolism causes JNK activation, which promotes mitochondrial dysfunction and results in the amplification of oxidative stress. Therefore, inhibition of the initial phase of JNK activation may be key to protection against APAP-induced liver injury. The goal of this study was to determine whether mixed-lineage kinase 3 (MLK3) mediates the initial, ASK1-independent phase of APAP-induced JNK activation and thus promotes drug-induced hepatotoxicity. We found that MLK3 was activated by oxidative stress and was required for JNK activation in response to oxidative stress. Loss of MLK3 attenuated APAP-induced JNK activation and hepatocyte death in vitro, independent of receptor-interacting protein 1. Moreover, JNK and glycogen synthase kinase 3β activation was significantly attenuated, and Mcl-1 degradation was inhibited in APAP-treated MLK3-knockout mice. Furthermore, we showed that loss of MLK3 increased expression of glutamate cysteine ligase, accelerated hepatic GSH recovery, and decreased production of reactive oxygen species after APAP treatment. MLK3-deficient mice were significantly protected from APAP-induced liver injury, compared with wild-type mice. Together, these studies establish a novel role for MLK3 in APAP-induced JNK activation and hepatotoxicity, and they suggest MLK3 as a possible target in the treatment of APAP-induced liver injury.
Figures
Similar articles
-
Inhibitor of apoptosis signal-regulating kinase 1 protects against acetaminophen-induced liver injury.Toxicol Appl Pharmacol. 2015 Jul 1;286(1):1-9. doi: 10.1016/j.taap.2015.03.019. Epub 2015 Mar 25. Toxicol Appl Pharmacol. 2015. PMID: 25818599 Free PMC article.
-
Silencing glycogen synthase kinase-3beta inhibits acetaminophen hepatotoxicity and attenuates JNK activation and loss of glutamate cysteine ligase and myeloid cell leukemia sequence 1.J Biol Chem. 2010 Mar 12;285(11):8244-55. doi: 10.1074/jbc.M109.054999. Epub 2010 Jan 8. J Biol Chem. 2010. PMID: 20061376 Free PMC article.
-
Protein tyrosine phosphatase 1B modulates GSK3β/Nrf2 and IGFIR signaling pathways in acetaminophen-induced hepatotoxicity.Cell Death Dis. 2013 May 9;4(5):e626. doi: 10.1038/cddis.2013.150. Cell Death Dis. 2013. PMID: 23661004 Free PMC article.
-
Pathophysiological significance of c-jun N-terminal kinase in acetaminophen hepatotoxicity.Expert Opin Drug Metab Toxicol. 2015;11(11):1769-79. doi: 10.1517/17425255.2015.1071353. Epub 2015 Jul 20. Expert Opin Drug Metab Toxicol. 2015. PMID: 26190663 Free PMC article. Review.
-
[c-Jun N-terminal kinase signaling pathway in acetaminophen-induced liver injury].Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 2023 Nov;35(11):1223-1228. doi: 10.3760/cma.j.cn121430-20221205-01060. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 2023. PMID: 37987136 Review. Chinese.
Cited by
-
MLK3 localizes mainly to the cytoplasm and promotes oxidative stress injury via a positive feedback loop.Cell Biochem Biophys. 2023 Sep;81(3):469-479. doi: 10.1007/s12013-023-01159-8. Epub 2023 Aug 7. Cell Biochem Biophys. 2023. PMID: 37550525
-
Mechanisms of acetaminophen-induced cell death in primary human hepatocytes.Toxicol Appl Pharmacol. 2014 Sep 15;279(3):266-274. doi: 10.1016/j.taap.2014.05.010. Epub 2014 Jun 3. Toxicol Appl Pharmacol. 2014. PMID: 24905542 Free PMC article.
-
Increased hepatic receptor interacting protein kinase 3 expression due to impaired proteasomal functions contributes to alcohol-induced steatosis and liver injury.Oncotarget. 2016 Apr 5;7(14):17681-98. doi: 10.18632/oncotarget.6893. Oncotarget. 2016. PMID: 26769846 Free PMC article.
-
Inhibitor of apoptosis signal-regulating kinase 1 protects against acetaminophen-induced liver injury.Toxicol Appl Pharmacol. 2015 Jul 1;286(1):1-9. doi: 10.1016/j.taap.2015.03.019. Epub 2015 Mar 25. Toxicol Appl Pharmacol. 2015. PMID: 25818599 Free PMC article.
-
Resveratrol prevents protein nitration and release of endonucleases from mitochondria during acetaminophen hepatotoxicity.Food Chem Toxicol. 2015 Jul;81:62-70. doi: 10.1016/j.fct.2015.04.014. Epub 2015 Apr 9. Food Chem Toxicol. 2015. PMID: 25865938 Free PMC article.
References
-
- Adams ML, Pierce RH, Vail ME, White CC, Tonge RP, Kavanagh TJ, Fausto N, Nelson SD, Bruschi SA. (2001) Enhanced acetaminophen hepatotoxicity in transgenic mice overexpressing BCL-2. Mol Pharmacol 60:907–915 - PubMed
-
- Botta D, Shi S, White CC, Dabrowski MJ, Keener CL, Srinouanprachanh SL, Farin FM, Ware CB, Ladiges WC, Pierce RH, et al. (2006) Acetaminophen-induced liver injury is attenuated in male glutamate-cysteine ligase transgenic mice. J Biol Chem 281:28865–28875 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
