

Evolutionary blueprint for host- and niche-adaptation in Staphylococcus aureus clonal complex CC30
- PMID: 22919639
- PMCID: PMC3417553
- DOI: 10.3389/fcimb.2012.00048
Evolutionary blueprint for host- and niche-adaptation in Staphylococcus aureus clonal complex CC30
Abstract
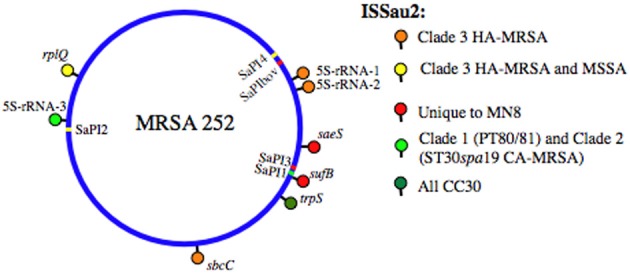
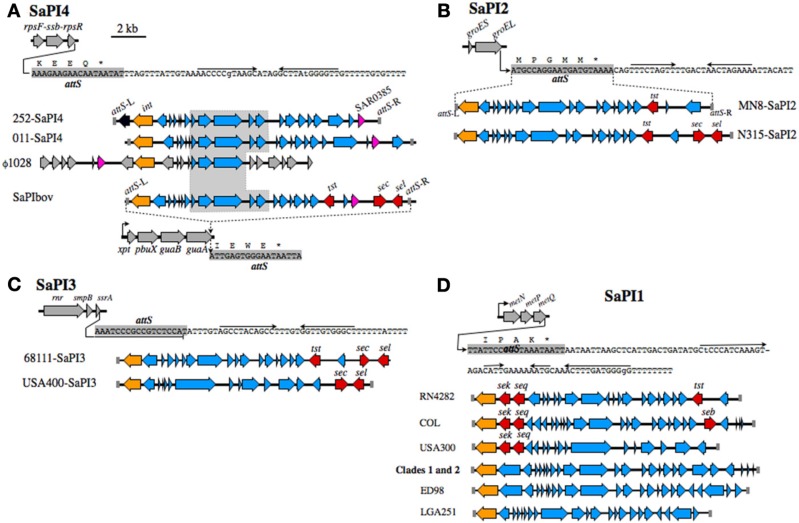
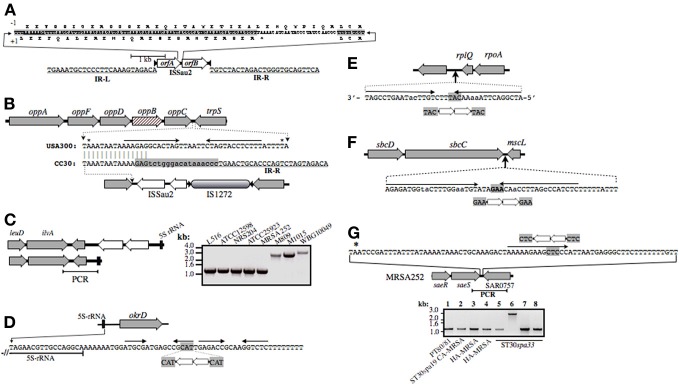
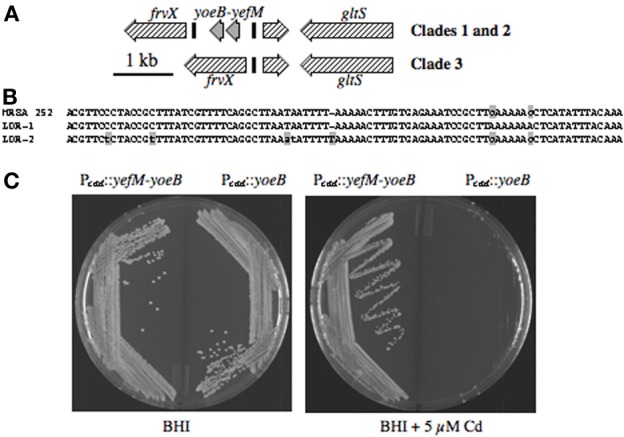
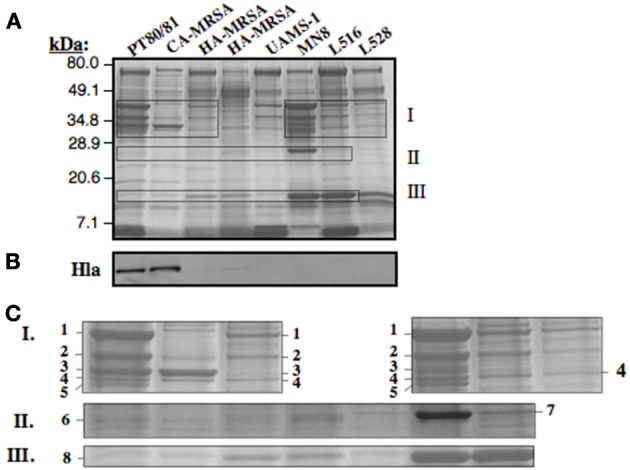
Staphylococcus aureus clonal complex CC30 has caused infectious epidemics for more than 60 years, and, therefore, provides a model system to evaluate how evolution has influenced the disease potential of closely related strains. In previous multiple genome comparisons, phylogenetic analyses established three major branches that evolved from a common ancestor. Clade 1, comprised of historic pandemic phage type 80/81 methicillin susceptible S. aureus (MSSA), and Clade 2 comprised of contemporary community acquired methicillin resistant S. aureus (CA-MRSA) were hyper-virulent in murine infection models. Conversely, Clade 3 strains comprised of contemporary hospital associated MRSA (HA-MRSA) and clinical MSSA exhibited attenuated virulence, due to common single nucleotide polymorphisms (SNP's) that abrogate production of α-hemolysin Hla, and interfere with signaling of the accessory gene regulator agr. We have now completed additional in silico genome comparisons of 15 additional CC30 genomes in the public domain, to assess the hypothesis that Clade 3 has evolved to favor niche adaptation. In addition to SNP's that influence agr and hla, other common traits of Clade 3 include tryptophan auxotrophy due to a di-nucleotide deletion within trpD, a premature stop codon within isdH encoding an immunogenic cell surface protein involved in iron acquisition, loss of a genomic toxin-antitoxin (TA) addiction module, acquisition of S. aureus pathogenicity islands SaPI4, and SaPI2 encoding toxic shock syndrome toxin tst, and increased copy number of insertion sequence ISSau2, which appears to target transcription terminators. Compared to other Clade 3 MSSA, S. aureus MN8, which is associated with Staphylococcal toxic shock syndrome, exhibited a unique ISSau2 insertion, and enhanced production of toxic shock syndrome toxin encoded by SaPI2. Cumulatively, our data support the notion that Clade 3 strains are following an evolutionary blueprint toward niche-adaptation.
Keywords: Staphylococcus aureus; evolution; insertion sequence; pathoadaptation; pathogenicity island; pseudogene; toxin-antitoxin addiction module; virulence.
Figures
Similar articles
-
Staphylococcus aureus nasal carriage, virulence traits, antibiotic resistance mechanisms, and genetic lineages in healthy humans in Spain, with detection of CC398 and CC97 strains.Int J Med Microbiol. 2011 Aug;301(6):500-5. doi: 10.1016/j.ijmm.2011.02.004. Epub 2011 May 12. Int J Med Microbiol. 2011. PMID: 21570348
-
Molecular differentiation of historic phage-type 80/81 and contemporary epidemic Staphylococcus aureus.Proc Natl Acad Sci U S A. 2011 Nov 1;108(44):18091-6. doi: 10.1073/pnas.1111084108. Epub 2011 Oct 24. Proc Natl Acad Sci U S A. 2011. PMID: 22025717 Free PMC article.
-
Healthcare- and Community-Associated Methicillin-Resistant Staphylococcus aureus (MRSA) and Fatal Pneumonia with Pediatric Deaths in Krasnoyarsk, Siberian Russia: Unique MRSA's Multiple Virulence Factors, Genome, and Stepwise Evolution.PLoS One. 2015 Jun 5;10(6):e0128017. doi: 10.1371/journal.pone.0128017. eCollection 2015. PLoS One. 2015. PMID: 26047024 Free PMC article.
-
Evolution and pathogenesis of Staphylococcus aureus: lessons learned from genotyping and comparative genomics.FEMS Microbiol Rev. 2008 Jan;32(1):23-37. doi: 10.1111/j.1574-6976.2007.00086.x. Epub 2007 Nov 5. FEMS Microbiol Rev. 2008. PMID: 17983441 Review.
-
Genomics of Natural Populations of Staphylococcus aureus.Annu Rev Microbiol. 2016 Sep 8;70:459-78. doi: 10.1146/annurev-micro-102215-095547. Epub 2016 Jul 29. Annu Rev Microbiol. 2016. PMID: 27482738 Review.
Cited by
-
Mutation of Agr Is Associated with the Adaptation of Staphylococcus aureus to the Host during Chronic Osteomyelitis.Front Cell Infect Microbiol. 2018 Feb 2;8:18. doi: 10.3389/fcimb.2018.00018. eCollection 2018. Front Cell Infect Microbiol. 2018. PMID: 29456969 Free PMC article.
-
Molecular Characterization of a Prevalent Ribocluster of Methicillin-Sensitive Staphylococcus aureus from Orthopedic Implant Infections. Correspondence with MLST CC30.Front Cell Infect Microbiol. 2016 Feb 16;6:8. doi: 10.3389/fcimb.2016.00008. eCollection 2016. Front Cell Infect Microbiol. 2016. PMID: 26909340 Free PMC article.
-
Distribution of Virulence Factors and Resistance Determinants in Three Genotypes of Staphylococcus argenteus Clinical Isolates in Japan.Pathogens. 2021 Feb 3;10(2):163. doi: 10.3390/pathogens10020163. Pathogens. 2021. PMID: 33546443 Free PMC article.
-
Decoding the evolutionary history of ST30 Staphylococcus aureus: insights into a potentially silent MSSA bloodstream pathogen.Front Microbiol. 2025 Apr 9;16:1522747. doi: 10.3389/fmicb.2025.1522747. eCollection 2025. Front Microbiol. 2025. PMID: 40270815 Free PMC article.
-
Potential Influence of Staphylococcus aureus Clonal Complex 30 Genotype and Transcriptome on Hematogenous Infections.Open Forum Infect Dis. 2015 Jun 24;2(3):ofv093. doi: 10.1093/ofid/ofv093. eCollection 2015 Sep. Open Forum Infect Dis. 2015. PMID: 26213692 Free PMC article.
References
-
- Altemeier W. A., Lewis S., Schlievert P. M., Bjornson H. S. (1981). Studies of the Staphylococcal causation of toxic shock syndrome. Surg. Gynecol. Obstet. 153, 481–485 - PubMed
-
- Baba T., Takeuchi F., Kuroda M., Yuzawa H., Aoki K., Oguchi A., Nagai Y., Iwama N., Asano K., Naimi T., Kuroda H., Cui L., Yamamoto K., Hiramatsu K. (2002). Genome and virulence determinants of high virulence community-acquired MRSA. Lancet 359, 1819–1827 10.1016/S0140-6736(02)08713-5 - DOI - PubMed
-
- Brazier J. S. (2008). Clostridium difficile: from obscurity to superbug. Br. J. Biomed. Sci. 65, 39–44 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials