

Yellow lupin (Lupinus luteus L.) transcriptome sequencing: molecular marker development and comparative studies
- PMID: 22920992
- PMCID: PMC3472298
- DOI: 10.1186/1471-2164-13-425
Yellow lupin (Lupinus luteus L.) transcriptome sequencing: molecular marker development and comparative studies
Abstract
Background: Yellow lupin (Lupinus luteus L.) is a minor legume crop characterized by its high seed protein content. Although grown in several temperate countries, its orphan condition has limited the generation of genomic tools to aid breeding efforts to improve yield and nutritional quality. In this study, we report the construction of 454-expresed sequence tag (EST) libraries, carried out comparative studies between L. luteus and model legume species, developed a comprehensive set of EST-simple sequence repeat (SSR) markers, and validated their utility on diversity studies and transferability to related species.
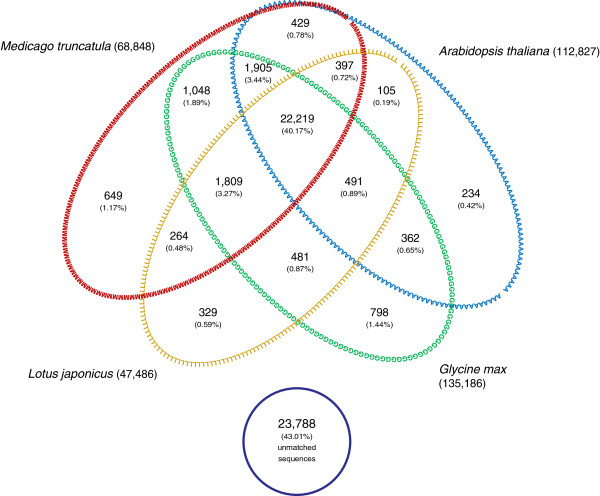
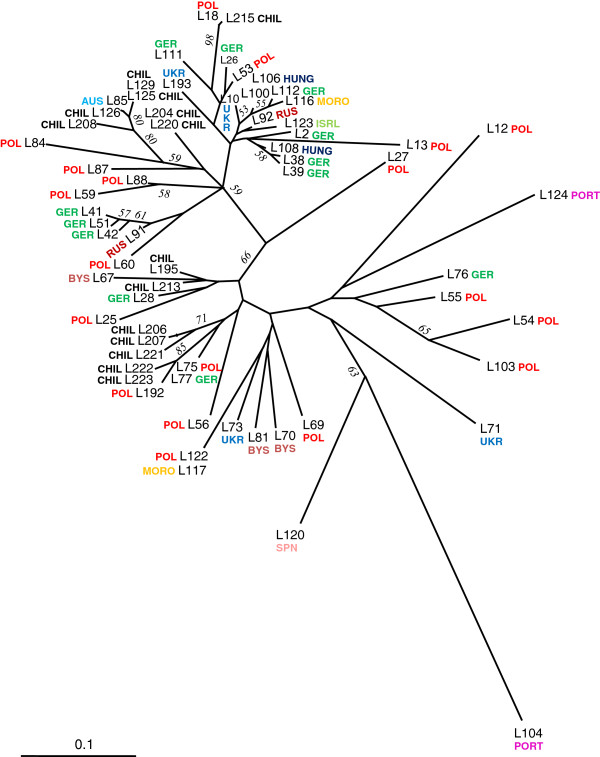
Results: Two runs of 454 pyrosequencing yielded 205 Mb and 530 Mb of sequence data for L1 (young leaves, buds and flowers) and L2 (immature seeds) EST- libraries. A combined assembly (L1L2) yielded 71,655 contigs with an average contig length of 632 nucleotides. L1L2 contigs were clustered into 55,309 isotigs. 38,200 isotigs translated into proteins and 8,741 of them were full length. Around 57% of L. luteus sequences had significant similarity with at least one sequence of Medicago, Lotus, Arabidopsis, or Glycine, and 40.17% showed positive matches with all of these species. L. luteus isotigs were also screened for the presence of SSR sequences. A total of 2,572 isotigs contained at least one EST-SSR, with a frequency of one SSR per 17.75 kbp. Empirical evaluation of the EST-SSR candidate markers resulted in 222 polymorphic EST-SSRs. Two hundred and fifty four (65.7%) and 113 (30%) SSR primer pairs were able to amplify fragments from L. hispanicus and L. mutabilis DNA, respectively. Fifty polymorphic EST-SSRs were used to genotype a sample of 64 L. luteus accessions. Neighbor-joining distance analysis detected the existence of several clusters among L. luteus accessions, strongly suggesting the existence of population subdivisions. However, no clear clustering patterns followed the accession's origin.
Conclusion: L. luteus deep transcriptome sequencing will facilitate the further development of genomic tools and lupin germplasm. Massive sequencing of cDNA libraries will continue to produce raw materials for gene discovery, identification of polymorphisms (SNPs, EST-SSRs, INDELs, etc.) for marker development, anchoring sequences for genome comparisons and putative gene candidates for QTL detection.
Figures



References
-
- Mace ES, Varshney RK, Mahalakshmi V, Seetha K, Gafoor A, Leeladevi Y, Crouch JH. In silico development of simple sequence repeat markers within the aeschynomenoid/dalbergoid and genistoid clades of the Leguminosae family and their transferability to Arachis hypogaea, groundnut. Plant Sci. 2008;174:51–60. doi: 10.1016/j.plantsci.2007.09.014. - DOI
-
- Gladstones JS. In: Lupins as crop plants: biology, production and utilization. Gladstones JS, Atkins CA, Hamblin J, editor. CAB International, Cambridge, United Kingdom; 1998. Distribution, origin, taxonomy, history and importance; pp. 1–37.
-
- Múzquiz M, Burbano C, Gorospe MJ, Ródenas I. A chemical study of Lupinus hispanicus seed—toxic and antinutritional components. J Scie Food Agr. 1989;47:205–214. doi: 10.1002/jsfa.2740470208. - DOI
-
- Wolko B, Weeden NF. Estimation of Lupinus genome polyploidy on the basis of isozymic loci number. Genet Pol. 1989;30:165–170.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
