

Mouse fukutin deletion impairs dystroglycan processing and recapitulates muscular dystrophy
- PMID: 22922256
- PMCID: PMC3428090
- DOI: 10.1172/JCI63004
Mouse fukutin deletion impairs dystroglycan processing and recapitulates muscular dystrophy
Abstract
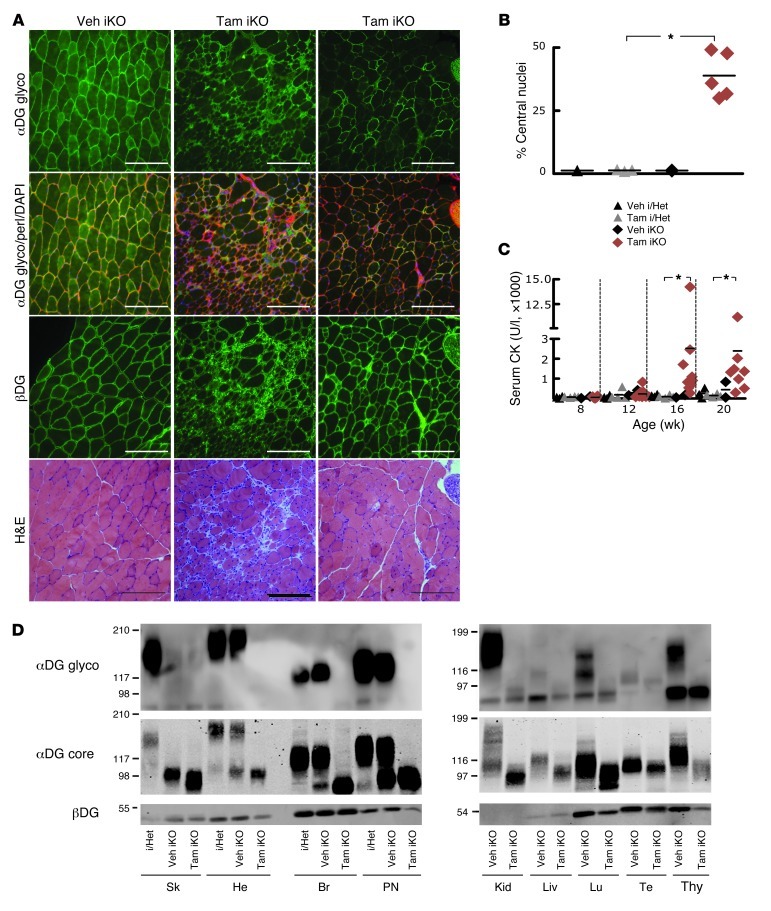
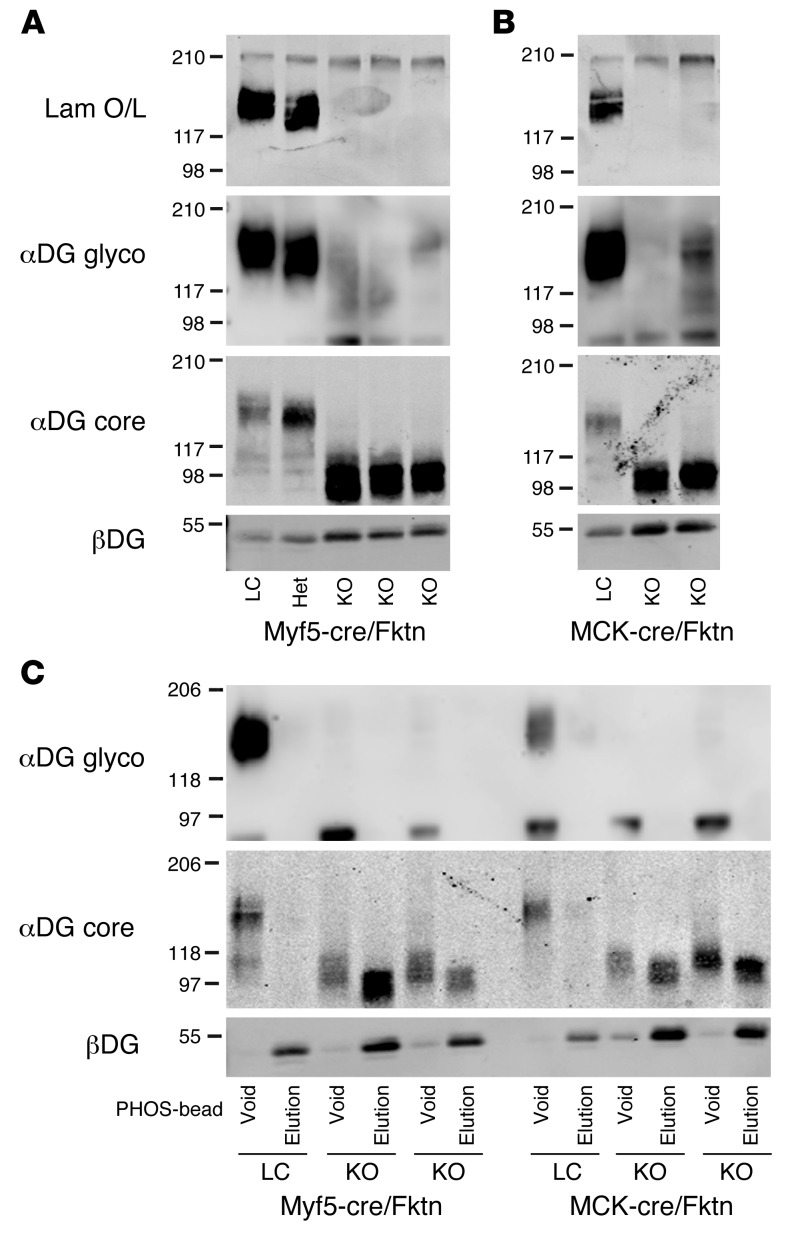
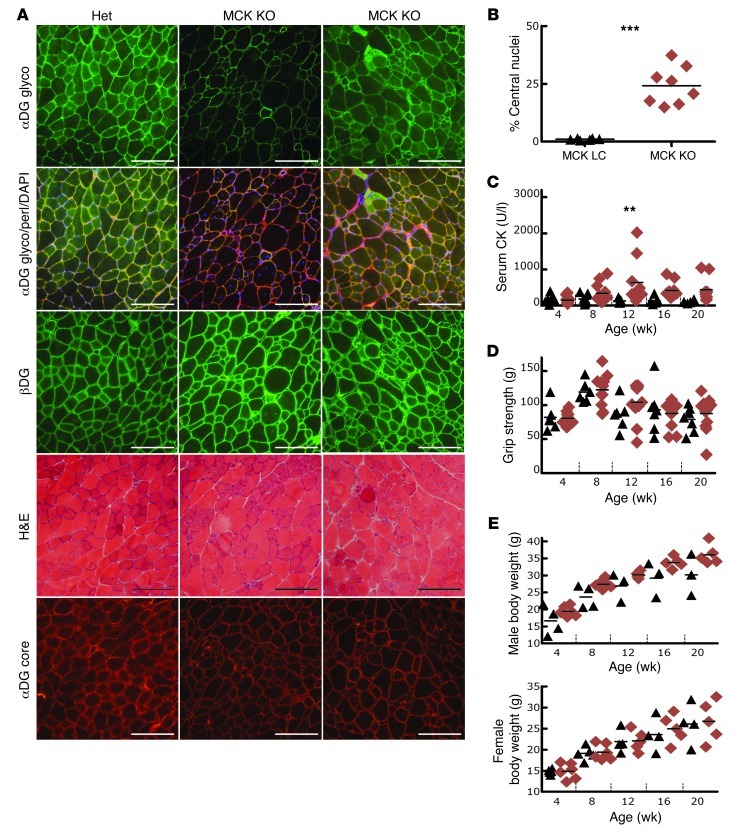
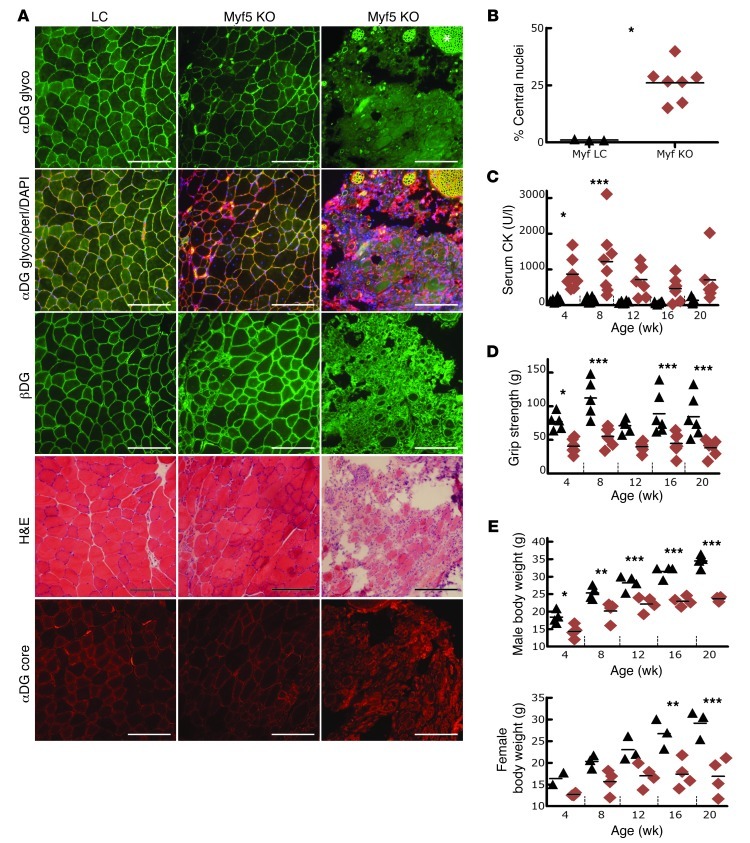
Dystroglycan is a transmembrane glycoprotein that links the extracellular basement membrane to cytoplasmic dystrophin. Disruption of the extensive carbohydrate structure normally present on α-dystroglycan causes an array of congenital and limb girdle muscular dystrophies known as dystroglycanopathies. The essential role of dystroglycan in development has hampered elucidation of the mechanisms underlying dystroglycanopathies. Here, we developed a dystroglycanopathy mouse model using inducible or muscle-specific promoters to conditionally disrupt fukutin (Fktn), a gene required for dystroglycan processing. In conditional Fktn-KO mice, we observed a near absence of functionally glycosylated dystroglycan within 18 days of gene deletion. Twenty-week-old KO mice showed clear dystrophic histopathology and a defect in glycosylation near the dystroglycan O-mannose phosphate, whether onset of Fktn excision driven by muscle-specific promoters occurred at E8 or E17. However, the earlier gene deletion resulted in more severe phenotypes, with a faster onset of damage and weakness, reduced weight and viability, and regenerating fibers of smaller size. The dependence of phenotype severity on the developmental timing of muscle Fktn deletion supports a role for dystroglycan in muscle development or differentiation. Moreover, given that this conditional Fktn-KO mouse allows the generation of tissue- and timing-specific defects in dystroglycan glycosylation, avoids embryonic lethality, and produces a phenotype resembling patient pathology, it is a promising new model for the study of secondary dystroglycanopathy.
Figures
Comment in
-
The attachment disorders of muscle: failure to carb-load.J Clin Invest. 2012 Sep;122(9):3046-8. doi: 10.1172/JCI65483. Epub 2012 Aug 27. J Clin Invest. 2012. PMID: 22922262 Free PMC article.
References
-
- Ervasti JM. Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim Biophys Acta. 2007;1772(2):108–117. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
