

Consistent neurodegeneration and its association with clinical progression in Huntington's disease: a coordinate-based meta-analysis
- PMID: 22922585
- PMCID: PMC4801478
- DOI: 10.1159/000339528
Consistent neurodegeneration and its association with clinical progression in Huntington's disease: a coordinate-based meta-analysis
Abstract
Background: The neuropathological hallmark of Huntington's disease (HD) is progressive striatal loss starting several years prior to clinical onset. In the past decade, whole-brain magnetic resonance imaging (MRI) studies have provided accumulating evidence for widely distributed cortical and subcortical atrophy in the early course of the disease.
Objective: In order to synthesize current morphometric MRI findings and to investigate the impact of clinical and genetic features on structural changes, we performed a coordinate-based meta-analysis of voxel-based morphometry (VBM) studies in HD.
Methods: Twenty HD samples derived from 17 studies were integrated in the analysis comparing a total of 685 HD mutation carriers [345 presymptomatic (pre-HD) and 340 symptomatic (symp-HD) subjects] and 507 controls. Convergent findings across studies were delineated using the anatomical likelihood estimation approach. Effects of genetic and clinical parameters on the likelihood of observing VBM findings were calculated by means of correlation analyses.
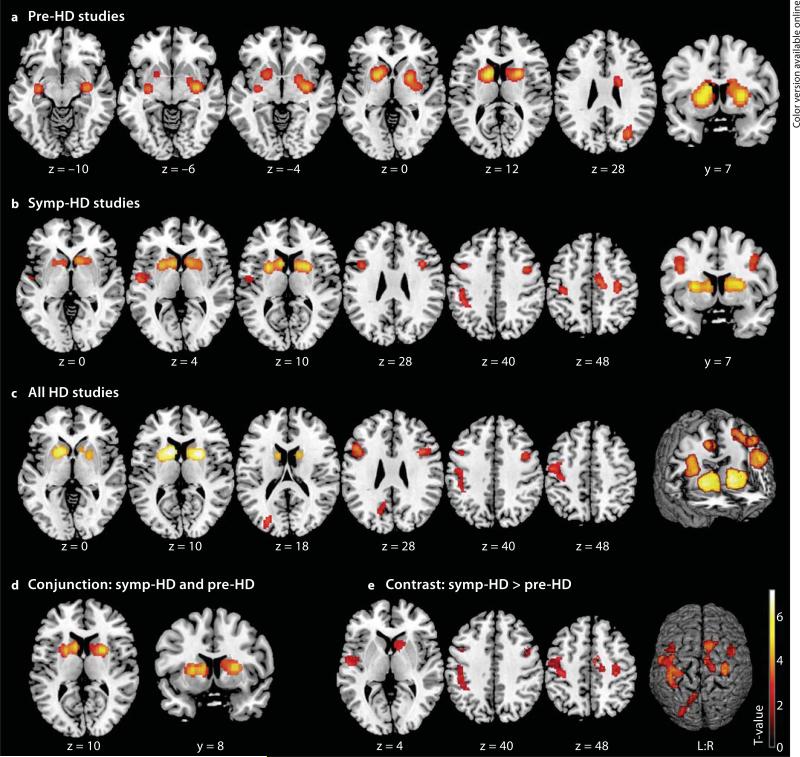
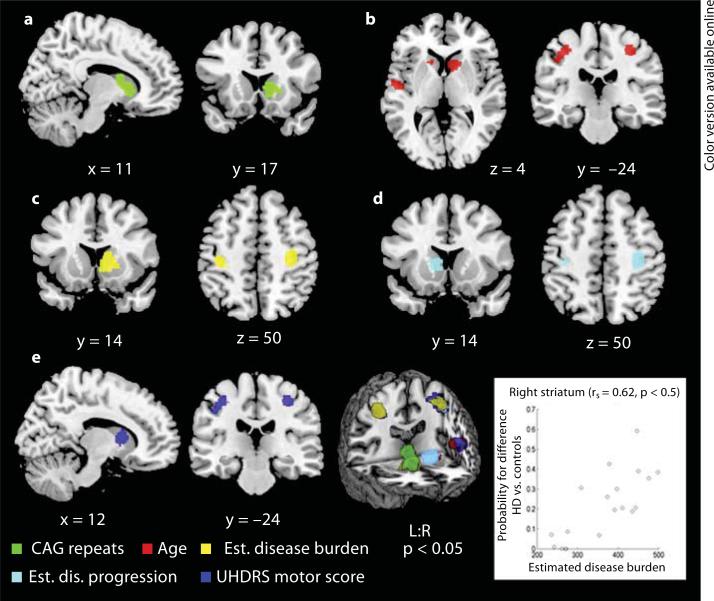
Results: Pre-HD studies featured convergent evidence for neurodegeneration in the basal ganglia, amygdala, thalamus, insula and occipital regions. In symp-HD, cerebral atrophy was more pronounced and spread to cortical regions (i.e., inferior frontal, premotor, sensorimotor, midcingulate, frontoparietal and temporoparietal cortices). Higher cytosine-adenosine-guanosine repeats were associated with striatal degeneration, while parameters of disease progression and motor impairment additionally correlated with cortical atrophy, especially in sensorimotor areas.
Conclusion: This first quantitative meta-analysis in HD demonstrates the extent of striatal atrophy and further consistent extrastriatal degeneration before clinical conversion. Sensorimotor areas seem to be core regions affected in symp-HD and, along with widespread cortical atrophy, may account for the clinical heterogeneity in HD.
Copyright © 2012 S. Karger AG, Basel.
Figures
References
-
- The Huntington's Disease Collaborative Research Group A novel gene containing a tri-nucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. - PubMed
-
- Aylward EH, Sparks BF, Field KM, Yallapragada V, Shpritz BD, Rosenblatt A, Brandt J, Gourley LM, Liang K, Zhou H, Margolis RL, Ross CA. Onset and rate of striatal atrophy in preclinical Huntington disease. Neurology. 2004;63:66–72. - PubMed
-
- Paulsen JS, Nopoulos PC, Aylward E, Ross CA, Johnson H, Magnotta VA, Juhl A, Pierson RK, Mills J, Langbehn D, Nance M. PREDICT-HD Investigators and Coordinators of the Huntington's Study Group (HSG): Striatal and white matter predictors of estimated diagnosis for Huntington disease. Brain Res Bull. 2010;82:201–207. - PMC - PubMed
-
- Bohanna I, Georgiou-Karistianis N, Hannan AJ, Egan GF. Magnetic resonance imaging as an approach towards identifying neuro-pathological biomarkers for Huntington's disease. Brain Res Rev. 2008;58:209–225. - PubMed
-
- Paulsen JS, Hayden M, Stout JC, Langbehn DR, Aylward E, Ross CA, Guttman M, Nance M, Kieburtz K, Oakes D, Shoulson I, Kayson E, Johnson S, Penziner E. Preparing for preventive clinical trials: the Predict-HD study. Arch Neurol. 2006;63:883–890. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
