

Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury
- PMID: 22922784
- PMCID: PMC3697869
- DOI: 10.1038/nn.3195
Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury
Abstract
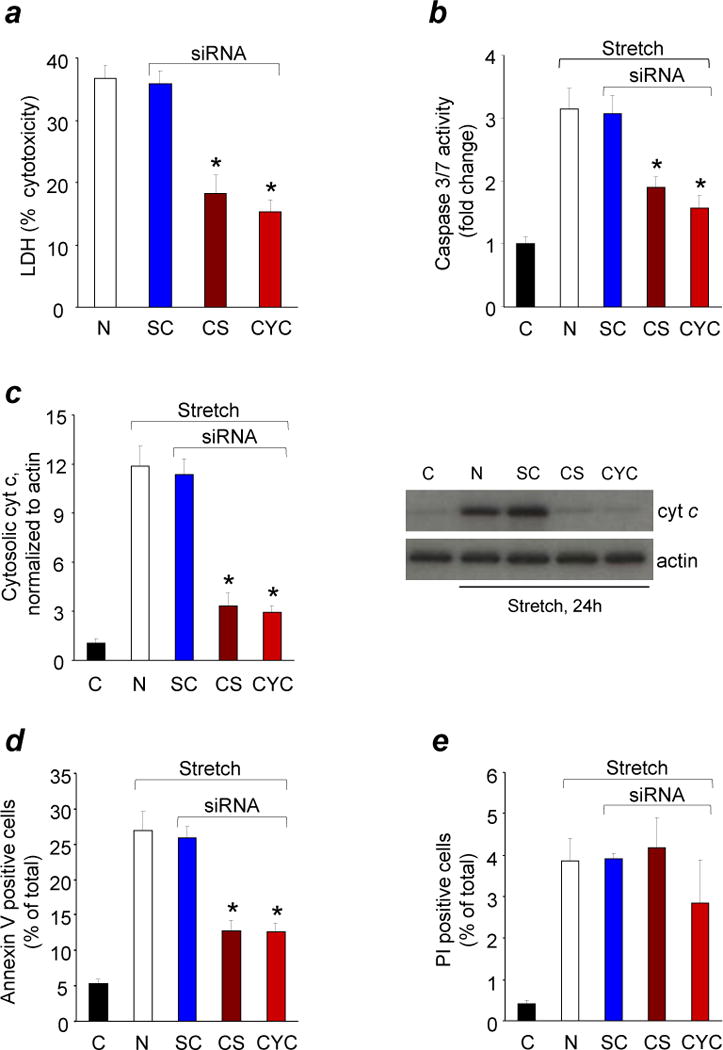
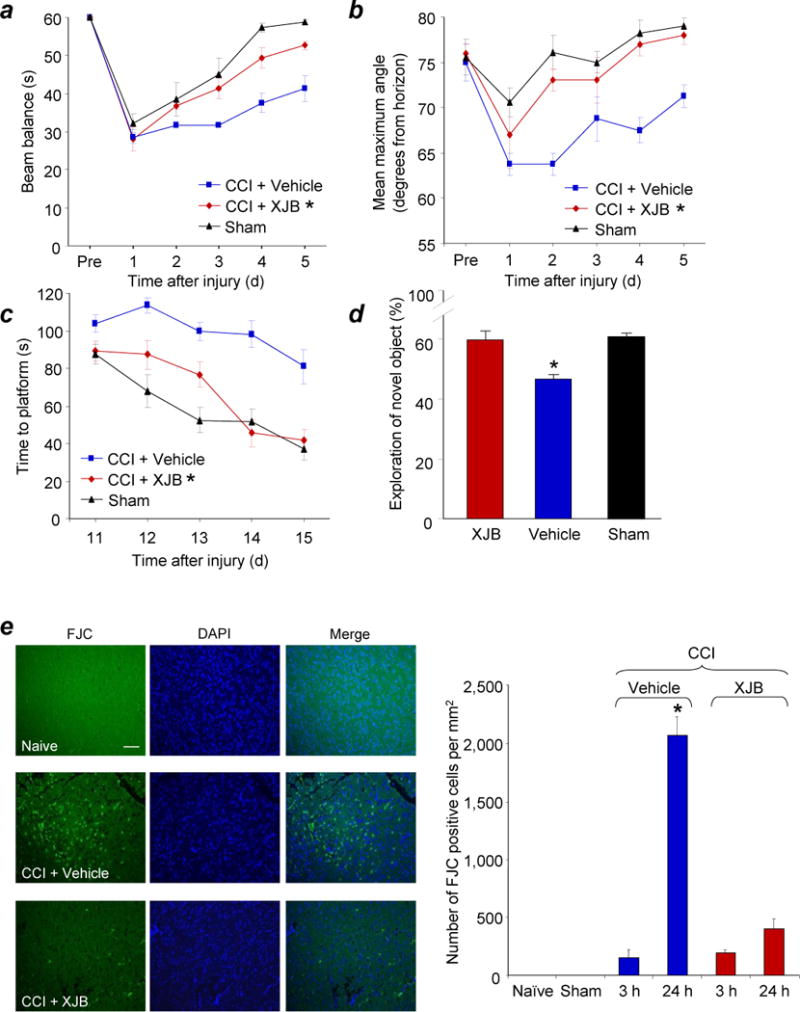
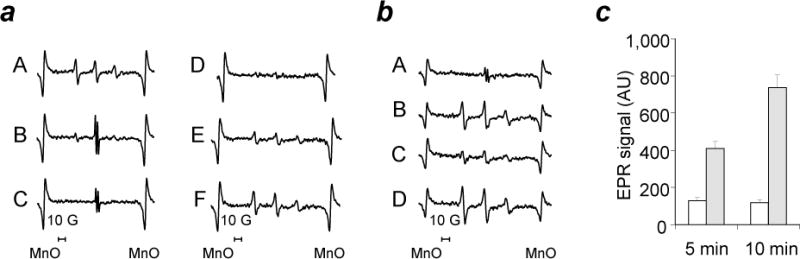
The brain contains a highly diversified complement of molecular species of a mitochondria-specific phospholipid, cardiolipin, which, because of its polyunsaturation, can readily undergo oxygenation. Using global lipidomics analysis in experimental traumatic brain injury (TBI), we found that TBI was accompanied by oxidative consumption of polyunsaturated cardiolipin and the accumulation of more than 150 new oxygenated molecular species of cardiolipin. RNAi-based manipulations of cardiolipin synthase and cardiolipin levels conferred resistance to mechanical stretch, an in vitro model of traumatic neuronal injury, in primary rat cortical neurons. By applying a brain-permeable mitochondria-targeted electron scavenger, we prevented cardiolipin oxidation in the brain, achieved a substantial reduction in neuronal death both in vitro and in vivo, and markedly reduced behavioral deficits and cortical lesion volume. We conclude that cardiolipin oxygenation generates neuronal death signals and that prevention of it by mitochondria-targeted small molecule inhibitors represents a new target for neuro-drug discovery.
Figures




Comment in
-
Knockout punch: cardiolipin oxidation in trauma.Nat Neurosci. 2012 Oct;15(10):1325-7. doi: 10.1038/nn.3222. Nat Neurosci. 2012. PMID: 23007184 No abstract available.
References
-
- Faul M, Xu L, Wald MM, Coronado VG. Traumatic brain injury in the United States: emergency department visits, hospitalizations, and deaths 2002-2006. Atlanta (GA): Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; 2010.
-
- Thurman DJ, Alverson C, Dunn KA, Guerrero J, Sniezek JE. Traumatic brain injury in the United States: A public health perspective. J Head Trauma Rehabil. 1999;14:602–615. - PubMed
-
- RAND. RAND Invisible Wounds of War Study. 2011 Jun 26; www.rand.org/multi/military/veterans.html.
-
- Ghajar J. Traumatic brain injury. Lancet. 2000;356:923–929. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL070755/HL/NHLBI NIH HHS/United States
- R01 OH008282/OH/NIOSH CDC HHS/United States
- U19 AI068021/AI/NIAID NIH HHS/United States
- R37 NS061817/NS/NINDS NIH HHS/United States
- R01 NS076511/NS/NINDS NIH HHS/United States
- ES020693/ES/NIEHS NIH HHS/United States
- U19AI068021/AI/NIAID NIH HHS/United States
- HL070755/HL/NHLBI NIH HHS/United States
- NS060005/NS/NINDS NIH HHS/United States
- R01 NS060005/NS/NINDS NIH HHS/United States
- NS061817/NS/NINDS NIH HHS/United States
- R01 ES020693/ES/NIEHS NIH HHS/United States
- R01 HD069620/HD/NICHD NIH HHS/United States
- NS076511/NS/NINDS NIH HHS/United States
- OH008282/OH/NIOSH CDC HHS/United States
- R01 NS061817/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
