

Locating a plausible binding site for an open-channel blocker, GlyH-101, in the pore of the cystic fibrosis transmembrane conductance regulator
- PMID: 22923500
- PMCID: PMC3502623
- DOI: 10.1124/mol.112.080267
Locating a plausible binding site for an open-channel blocker, GlyH-101, in the pore of the cystic fibrosis transmembrane conductance regulator
Abstract
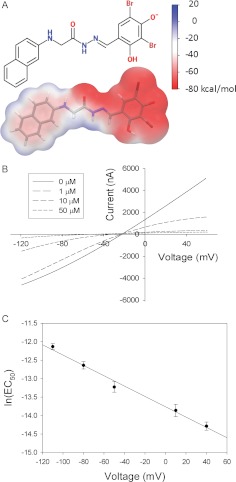
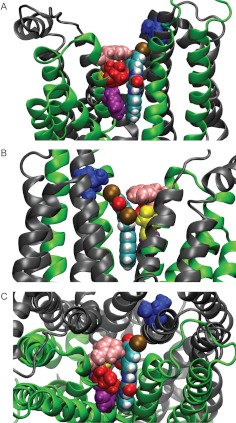
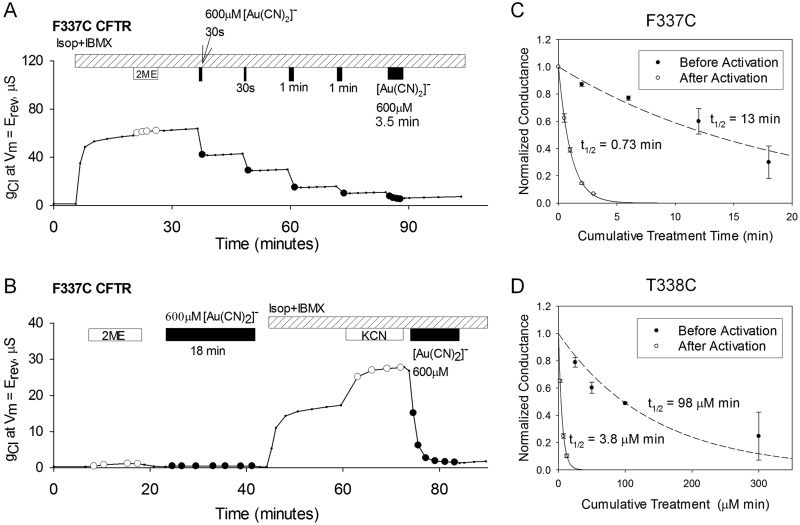
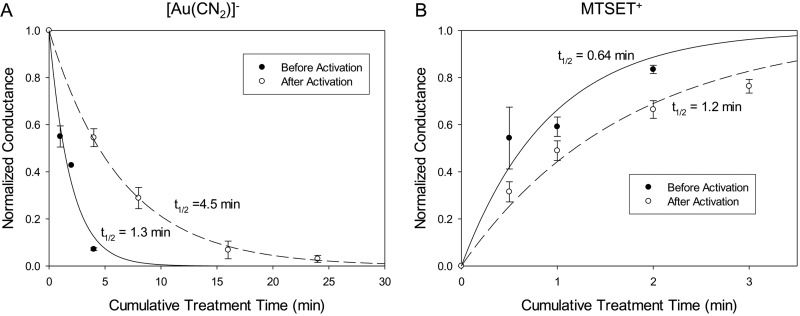
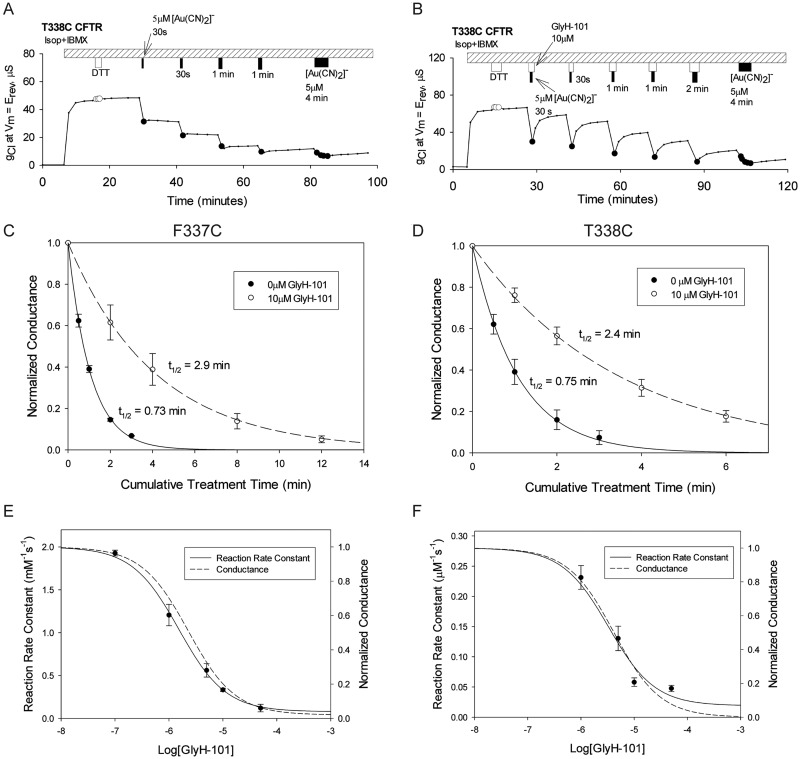
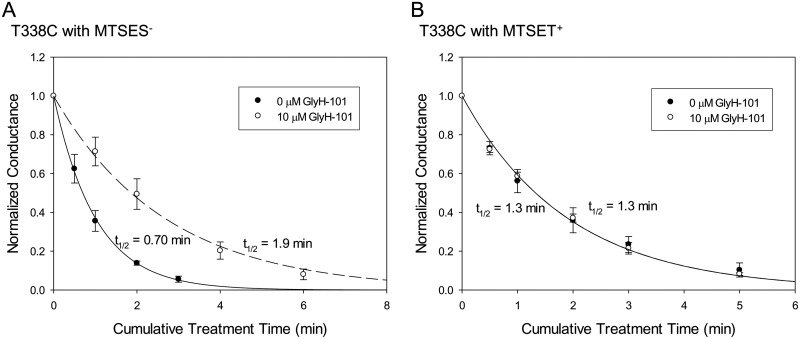
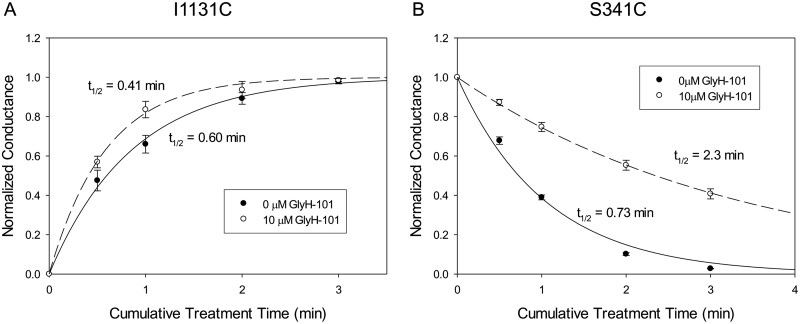
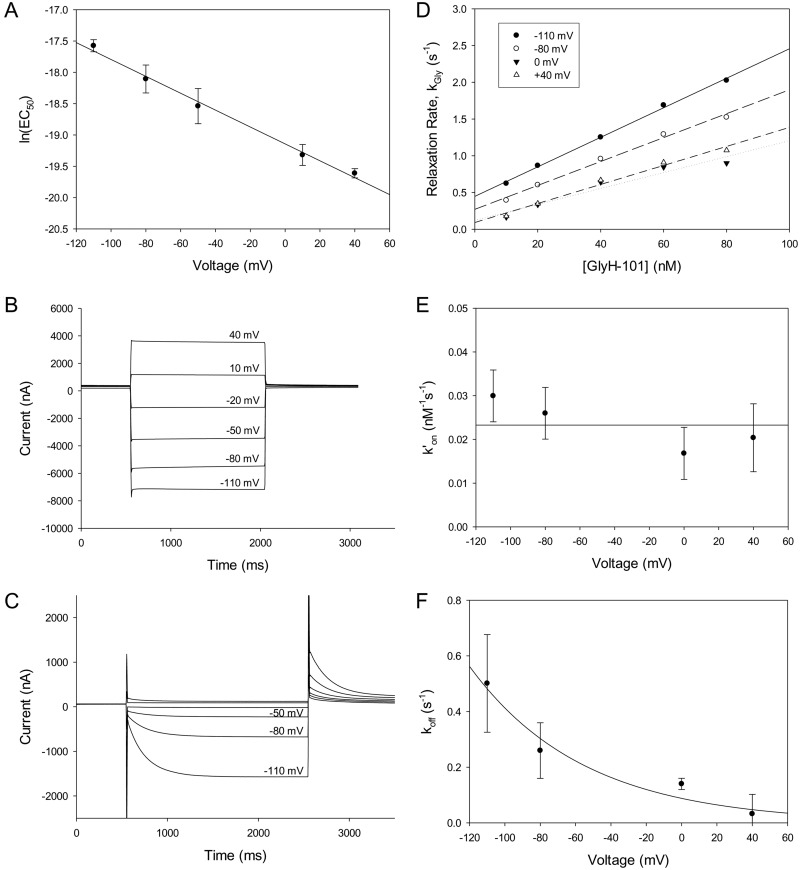
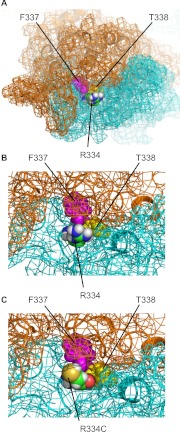
High-throughput screening has led to the identification of small-molecule blockers of the cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel, but the structural basis of blocker binding remains to be defined. We developed molecular models of the CFTR channel on the basis of homology to the bacterial transporter Sav1866, which could permit blocker binding to be analyzed in silico. The models accurately predicted the existence of a narrow region in the pore that is a likely candidate for the binding site of an open-channel pore blocker such as N-(2-naphthalenyl)-[(3,5-dibromo-2,4-dihydroxyphenyl)methylene]glycine hydrazide (GlyH-101), which is thought to act by entering the channel from the extracellular side. As a more-stringent test of predictions of the CFTR pore model, we applied induced-fit, virtual, ligand-docking techniques to identify potential binding sites for GlyH-101 within the CFTR pore. The highest-scoring docked position was near two pore-lining residues, Phe337 and Thr338, and the rates of reactions of anionic, thiol-directed reagents with cysteines substituted at these positions were slowed in the presence of the blocker, consistent with the predicted repulsive effect of the net negative charge on GlyH-101. When a bulky phenylalanine that forms part of the predicted binding pocket (Phe342) was replaced with alanine, the apparent affinity of the blocker was increased ∼200-fold. A molecular mechanics-generalized Born/surface area analysis of GlyH-101 binding predicted that substitution of Phe342 with alanine would substantially increase blocker affinity, primarily because of decreased intramolecular strain within the blocker-protein complex. This study suggests that GlyH-101 blocks the CFTR channel by binding within the pore bottleneck.
Figures
Similar articles
-
Discovery of glycine hydrazide pore-occluding CFTR inhibitors: mechanism, structure-activity analysis, and in vivo efficacy.J Gen Physiol. 2004 Aug;124(2):125-37. doi: 10.1085/jgp.200409059. J Gen Physiol. 2004. PMID: 15277574 Free PMC article.
-
Divergent CFTR orthologs respond differently to the channel inhibitors CFTRinh-172, glibenclamide, and GlyH-101.Am J Physiol Cell Physiol. 2012 Jan 1;302(1):C67-76. doi: 10.1152/ajpcell.00225.2011. Epub 2011 Sep 21. Am J Physiol Cell Physiol. 2012. PMID: 21940661 Free PMC article.
-
Cystic fibrosis transmembrane regulator inhibitors CFTR(inh)-172 and GlyH-101 target mitochondrial functions, independently of chloride channel inhibition.J Pharmacol Exp Ther. 2010 Apr;333(1):60-9. doi: 10.1124/jpet.109.162032. Epub 2010 Jan 5. J Pharmacol Exp Ther. 2010. PMID: 20051483
-
Therapeutic potential of cystic fibrosis transmembrane conductance regulator (CFTR) inhibitors in polycystic kidney disease.BioDrugs. 2009;23(4):203-16. doi: 10.2165/11313570-000000000-00000. BioDrugs. 2009. PMID: 19697963 Review.
-
Mechanism of chloride permeation in the cystic fibrosis transmembrane conductance regulator chloride channel.Exp Physiol. 2006 Jan;91(1):123-9. doi: 10.1113/expphysiol.2005.031757. Epub 2005 Sep 12. Exp Physiol. 2006. PMID: 16157656 Review.
Cited by
-
Cystic fibrosis transmembrane conductance regulator chloride channel blockers: Pharmacological, biophysical and physiological relevance.World J Biol Chem. 2014 Feb 26;5(1):26-39. doi: 10.4331/wjbc.v5.i1.26. World J Biol Chem. 2014. PMID: 24600512 Free PMC article. Review.
-
Benzopyrimido-pyrrolo-oxazine-dione (R)-BPO-27 Inhibits CFTR Chloride Channel Gating by Competition with ATP.Mol Pharmacol. 2015 Oct;88(4):689-96. doi: 10.1124/mol.115.098368. Epub 2015 Jul 14. Mol Pharmacol. 2015. PMID: 26174774 Free PMC article.
-
Localizing a gate in CFTR.Proc Natl Acad Sci U S A. 2015 Feb 24;112(8):2461-6. doi: 10.1073/pnas.1420676112. Epub 2015 Feb 9. Proc Natl Acad Sci U S A. 2015. PMID: 25675504 Free PMC article.
-
Residual function of cystic fibrosis mutants predicts response to small molecule CFTR modulators.JCI Insight. 2018 Jul 26;3(14):e121159. doi: 10.1172/jci.insight.121159. eCollection 2018 Jul 26. JCI Insight. 2018. PMID: 30046002 Free PMC article.
-
Mechanisms of CFTR functional variants that impair regulated bicarbonate permeation and increase risk for pancreatitis but not for cystic fibrosis.PLoS Genet. 2014 Jul 17;10(7):e1004376. doi: 10.1371/journal.pgen.1004376. eCollection 2014 Jul. PLoS Genet. 2014. PMID: 25033378 Free PMC article.
References
-
- Alexander C, Ivetac A, Liu X, Norimatsu Y, Serrano JR, Landstrom A, Sansom M, Dawson DC. (2009) Cystic fibrosis transmembrane conductance regulator: using differential reactivity toward channel-permeant and channel-impermeant thiol-reactive probes to test a molecular model for the pore. Biochemistry 48:10078–10088 - PMC - PubMed
-
- Beck EJ, Yang Y, Yaemsiri S, Raghuram V. (2008) Conformational changes in a pore-lining helix coupled to cystic fibrosis transmembrane conductance regulator channel gating. J Biol Chem 283:4957–4966 - PubMed
-
- Boucher RC. (2007) Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu Rev Med 58:157–170 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
