

Repeat or not repeat?--Statistical validation of tandem repeat prediction in genomic sequences
- PMID: 22923522
- PMCID: PMC3488214
- DOI: 10.1093/nar/gks726
Repeat or not repeat?--Statistical validation of tandem repeat prediction in genomic sequences
Abstract
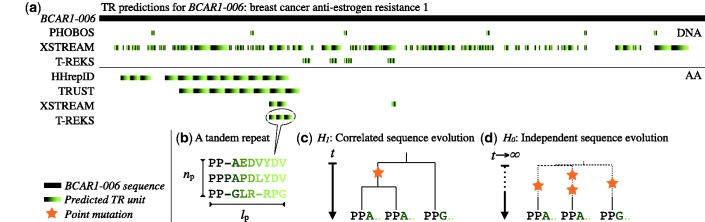
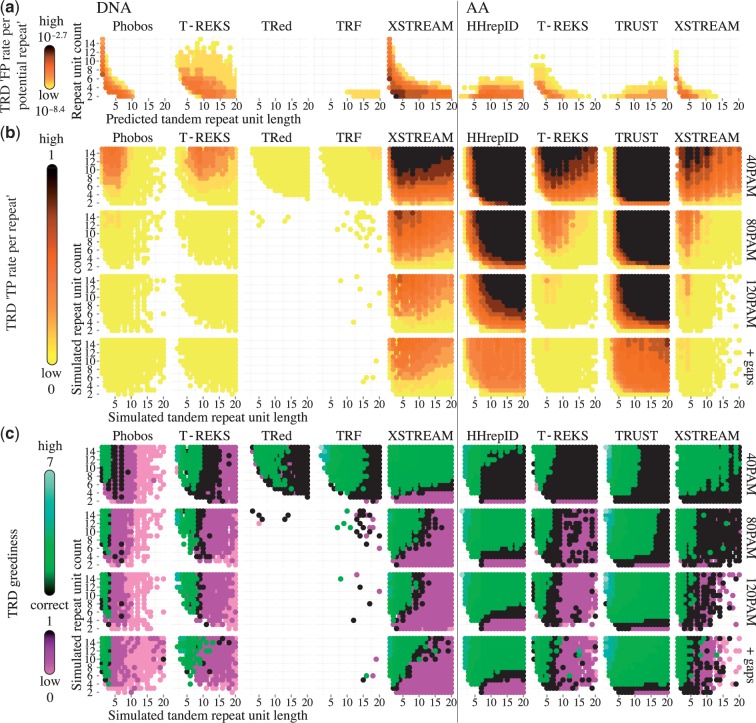
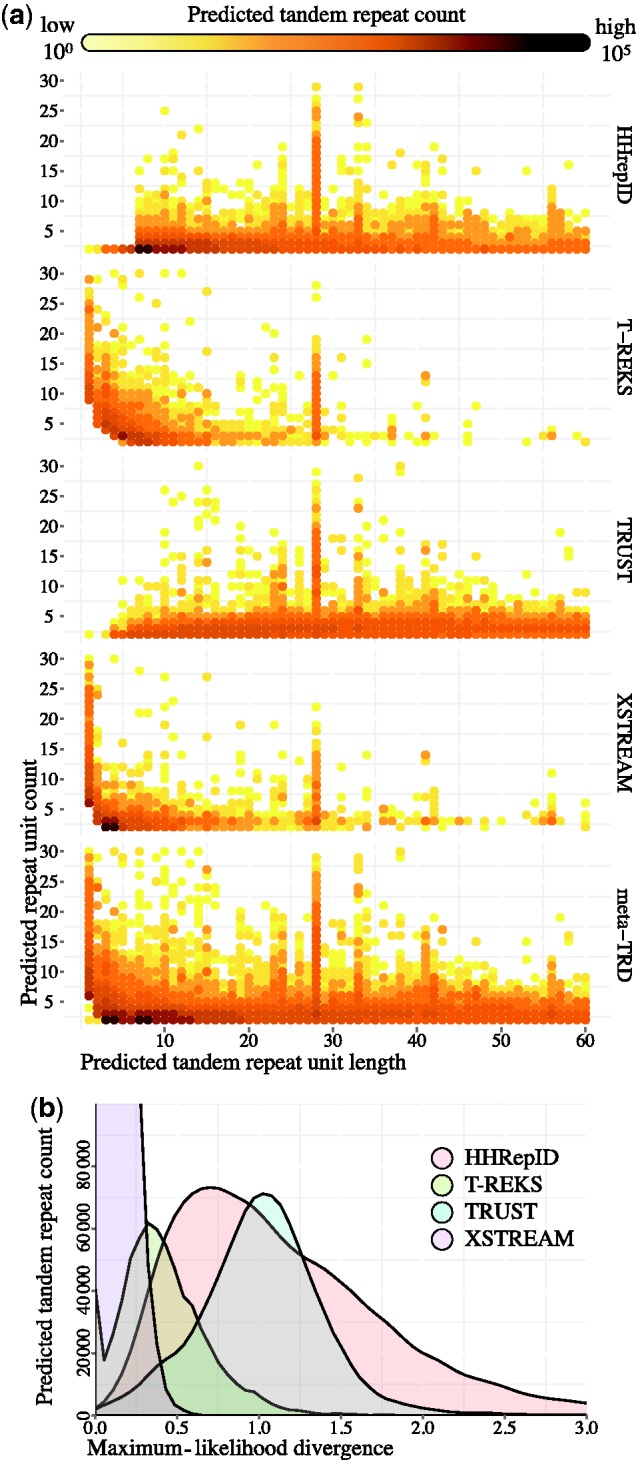
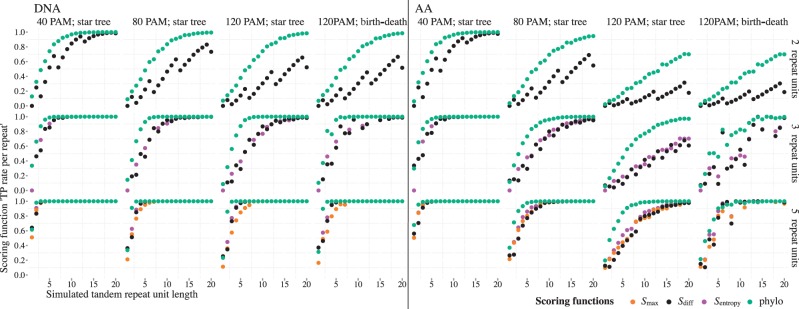
Tandem repeats (TRs) represent one of the most prevalent features of genomic sequences. Due to their abundance and functional significance, a plethora of detection tools has been devised over the last two decades. Despite the longstanding interest, TR detection is still not resolved. Our large-scale tests reveal that current detectors produce different, often nonoverlapping inferences, reflecting characteristics of the underlying algorithms rather than the true distribution of TRs in genomic data. Our simulations show that the power of detecting TRs depends on the degree of their divergence, and repeat characteristics such as the length of the minimal repeat unit and their number in tandem. To reconcile the diverse predictions of current algorithms, we propose and evaluate several statistical criteria for measuring the quality of predicted repeat units. In particular, we propose a model-based phylogenetic classifier, entailing a maximum-likelihood estimation of the repeat divergence. Applied in conjunction with the state of the art detectors, our statistical classification scheme for inferred repeats allows to filter out false-positive predictions. Since different algorithms appear to specialize at predicting TRs with certain properties, we advise applying multiple detectors with subsequent filtering to obtain the most complete set of genuine repeats.
Figures
References
-
- Jeffreys AJ, Wilson V, Thein SL. Individual-specific ‘fingerprints’ of human DNA. Nature. 1985;316:76–79. - PubMed
-
- Marcotte EM, Pellegrini M, Yeates TO, Eisenberg D. A census of protein repeats. J. Mol. Biol. 1999;293:151–160. - PubMed
-
- Itoh-Satoh M, Hayashi T, Nishi H, Koga Y, Arimura T, Koyanagi T, Takahashi M, Hohda S, Ueda K, Nouchi T, et al. Titin mutations as the molecular basis for dilated cardiomyopathy. Biochem. Biophy. Res. Commun. 2002;291:385–393. - PubMed
