

Reperfusion-induced myocardial dysfunction is prevented by endogenous annexin-A1 and its N-terminal-derived peptide Ac-ANX-A1(2-26)
- PMID: 22924634
- PMCID: PMC3570018
- DOI: 10.1111/j.1476-5381.2012.02176.x
Reperfusion-induced myocardial dysfunction is prevented by endogenous annexin-A1 and its N-terminal-derived peptide Ac-ANX-A1(2-26)
Abstract
Background and purpose: Annexin-A1 (ANX-A1) is an endogenous, glucocorticoid-regulated anti-inflammatory protein. The N-terminal-derived peptide Ac-ANX-A1(2-26) preserves cardiomyocyte viability, but the impact of ANX-A1-peptides on cardiac contractility is unknown. We now test the hypothesis that ANX-A1 preserves post-ischaemic recovery of left ventricular (LV) function.
Experimental approach: Ac-ANX-A1(2-26) was administered on reperfusion, to adult rat cardiomyocytes as well as hearts isolated from rats, wild-type mice and mice deficient in endogenous ANX-A1 (ANX-A1(-/-)). Myocardial viability and recovery of LV function were determined.
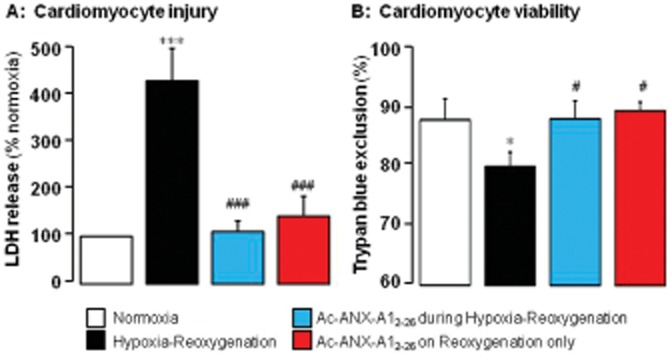
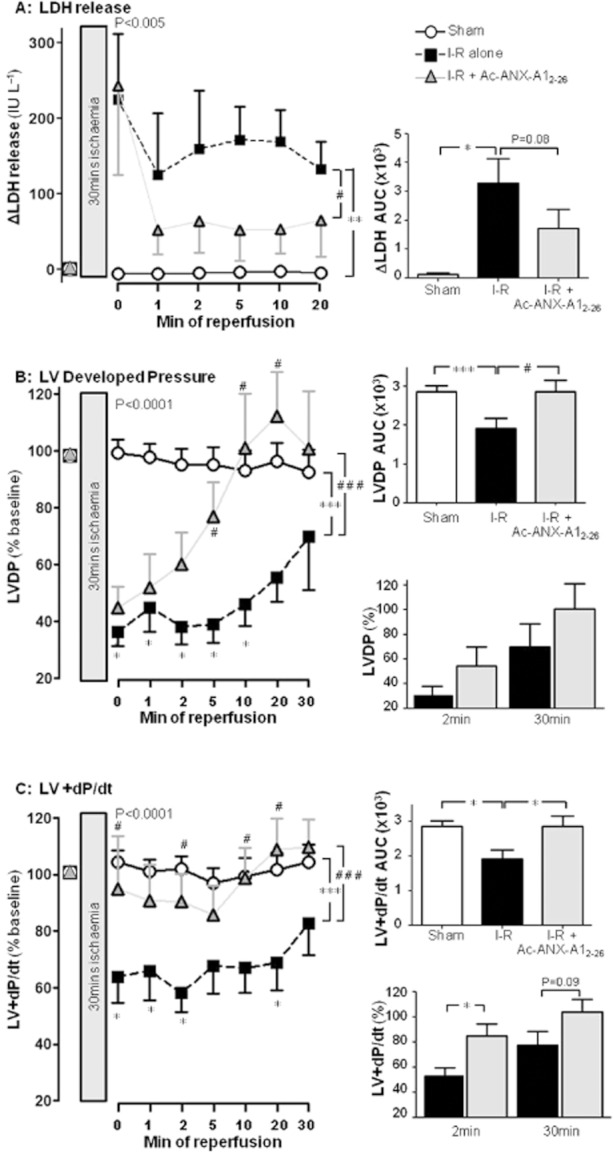
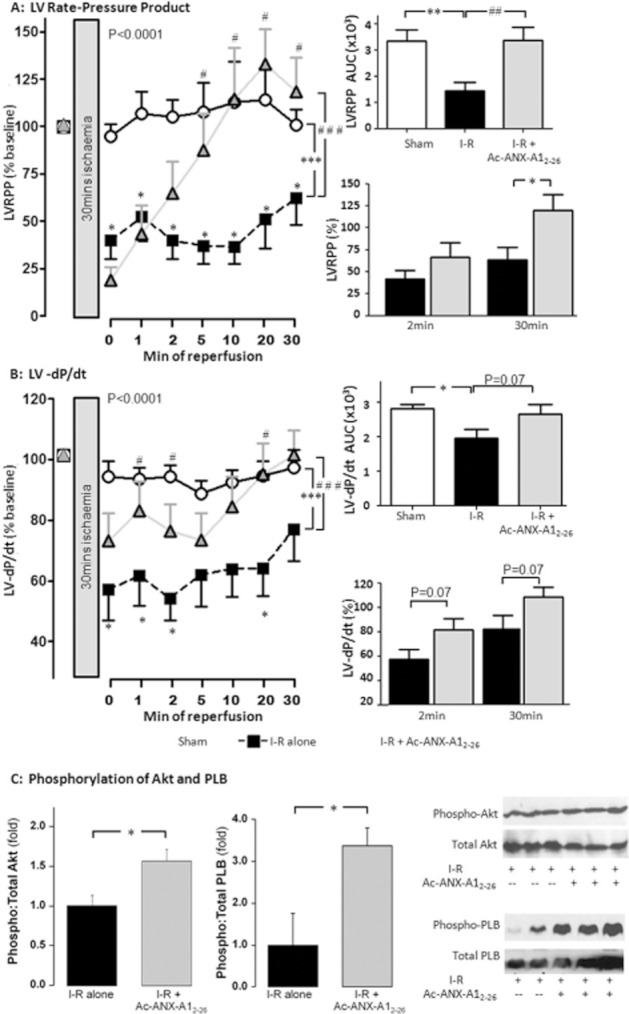
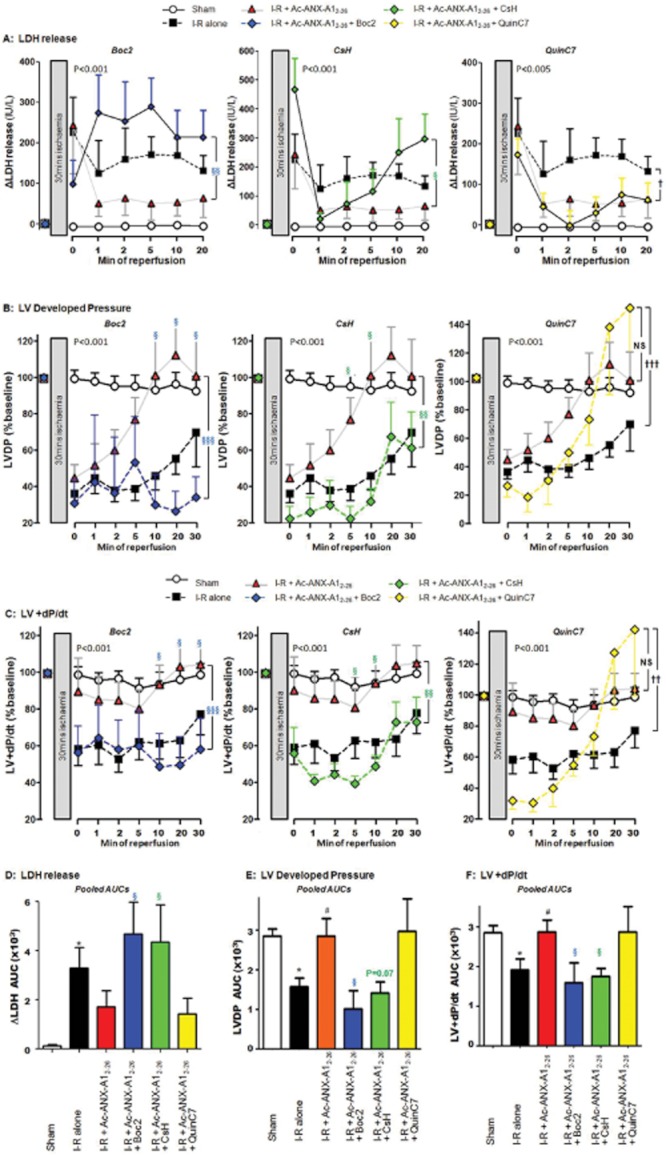
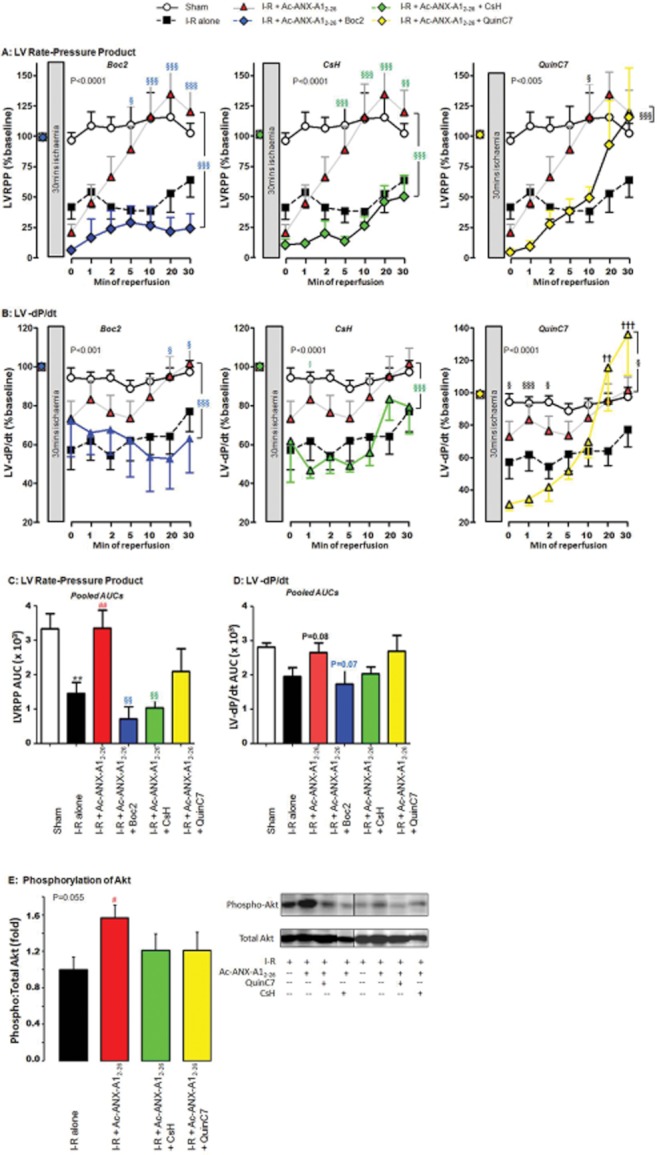
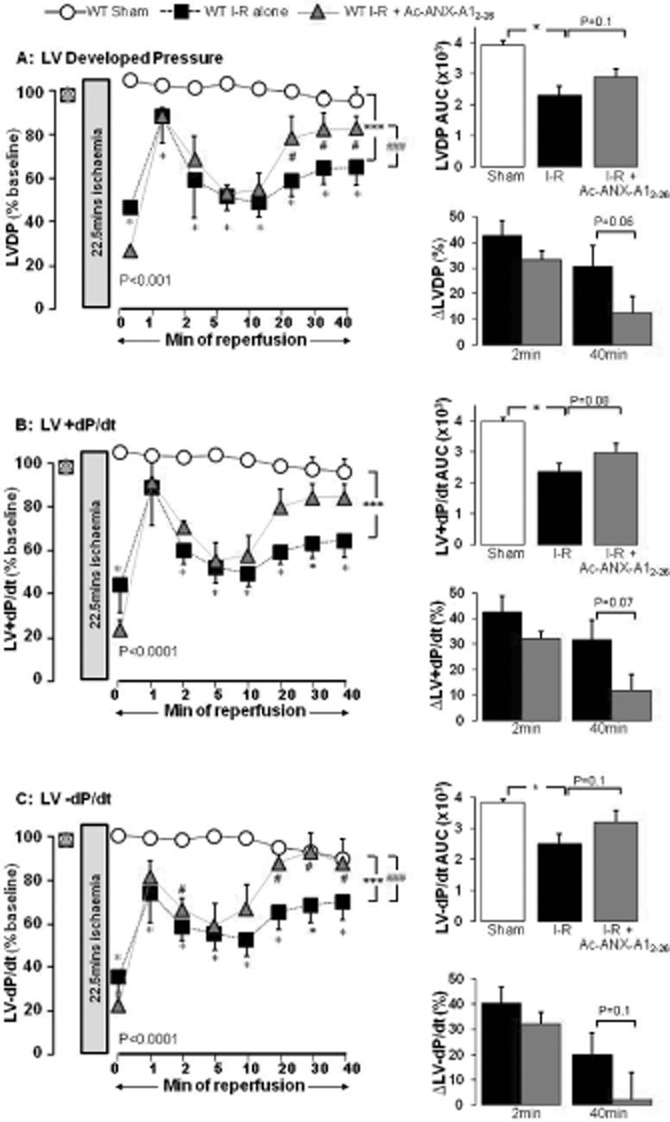
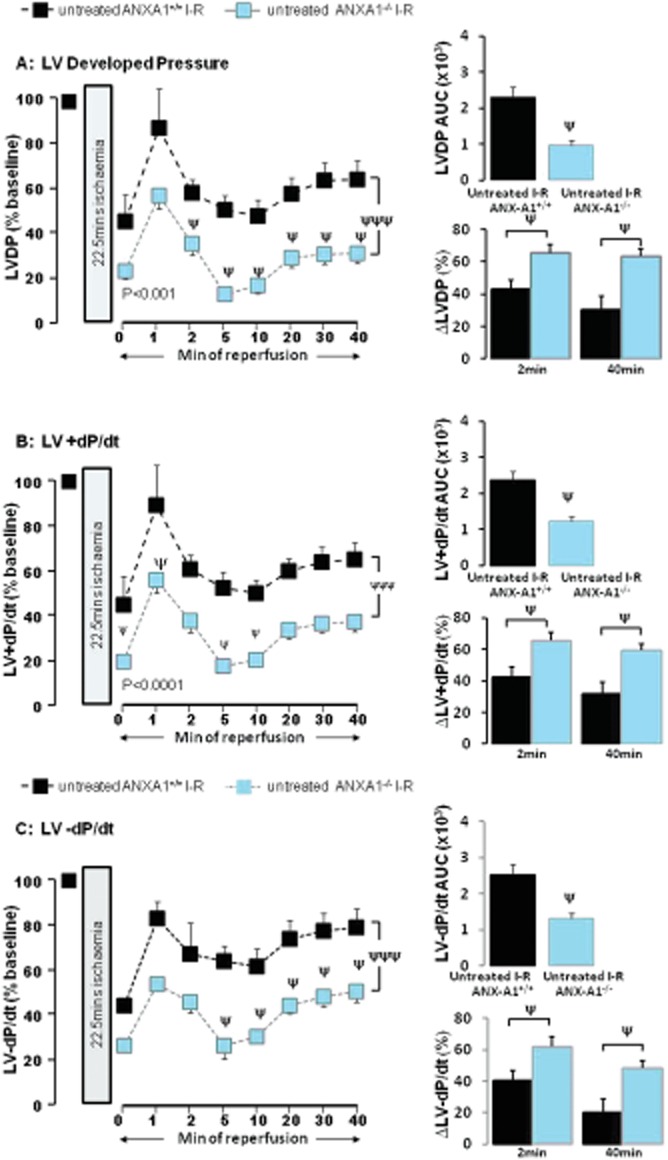
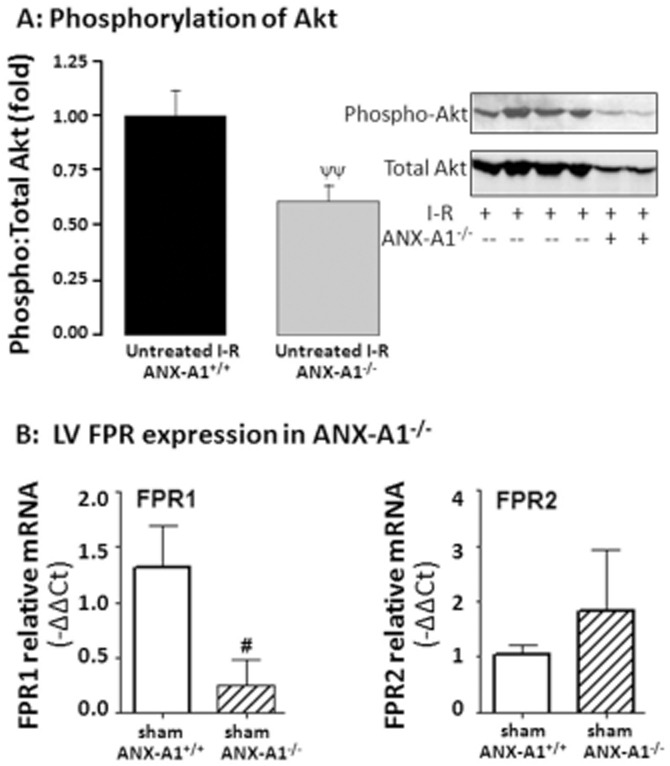
Key results: Ischaemia-reperfusion markedly impaired both cardiomyocyte viability and recovery of LV function by 60%. Treatment with exogenous Ac-ANX-A1(2-26) at the onset of reperfusion prevented cardiomyocyte injury and significantly improved recovery of LV function, in both intact rat and wild-type mouse hearts. Ac-ANX-A1(2-26) cardioprotection was abolished by either formyl peptide receptor (FPR)-nonselective or FPR1-selective antagonists, Boc2 and cyclosporin H, but was relatively insensitive to the FPR2-selective antagonist QuinC7. ANX-A1-induced cardioprotection was associated with increased phosphorylation of the cell survival kinase Akt. ANX-A1(-/-) exaggerated impairment of post-ischaemic recovery of LV function, in addition to selective LV FPR1 down-regulation.
Conclusions and implications: These data represent the first evidence that ANX-A1 affects myocardial function. Our findings suggest ANX-A1 is an endogenous regulator of post-ischaemic recovery of LV function. Furthermore, the ANX-A1-derived peptide Ac-ANX-A1(2-26) on reperfusion rescues LV function, probably via activation of FPR1. ANX-A1-based therapies may thus represent a novel clinical approach for the prevention and treatment of myocardial reperfusion injury.
© 2012 The Authors. British Journal of Pharmacology © 2012 The British Pharmacological Society.
Figures
References
-
- Burli RW, Xu H, Zou XM, Muller K, Golden J, Frohn M, et al. Potent hFPRL1 (ALXR) agonists as potential anti-inflammatory agents. Bioorg Med Chem Lett. 2006;16:3713–3718. - PubMed
-
- D'Amico M, Di Filippo C, La M, Solito E, McLean PG, Flower RJ, et al. Lipocortin 1 reduces myocardial ischemia-reperfusion injury by affecting local leukocyte recruitment. FASEB J. 2000;14:1867–1869. - PubMed
-
- Dreier R, Schmid KW, Gerke V, Riehemann K. Differential expression of annexins I, II and IV in human tissues: an immunohistochemical study. Histochem Cell Biol. 1998;110:137–148. - PubMed
-
- Dufton N, Perretti M. Therapeutic anti-inflammatory potential of formyl-peptide receptor agonists. Pharmacol Ther. 2010;127:175–188. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
