

Romidepsin (FK228) and its analogs directly inhibit phosphatidylinositol 3-kinase activity and potently induce apoptosis as histone deacetylase/phosphatidylinositol 3-kinase dual inhibitors
- PMID: 22924958
- PMCID: PMC7659383
- DOI: 10.1111/cas.12002
Romidepsin (FK228) and its analogs directly inhibit phosphatidylinositol 3-kinase activity and potently induce apoptosis as histone deacetylase/phosphatidylinositol 3-kinase dual inhibitors
Abstract
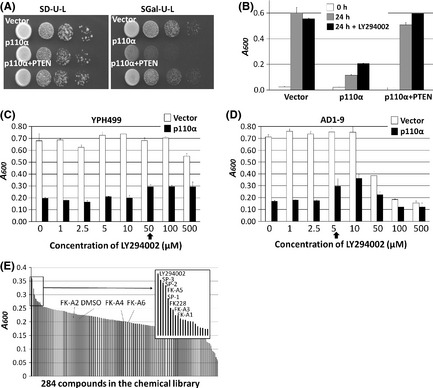
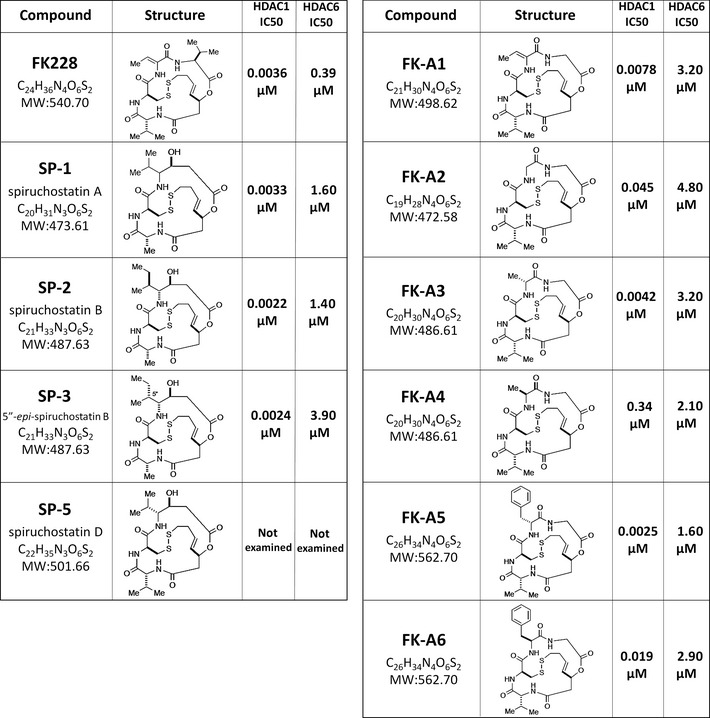
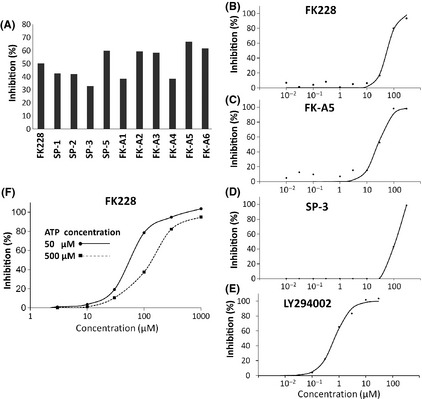
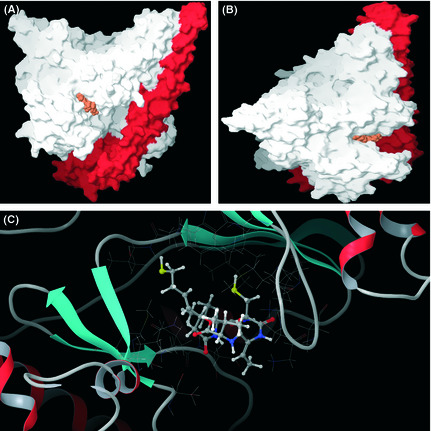
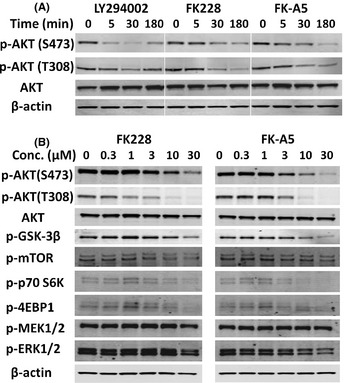
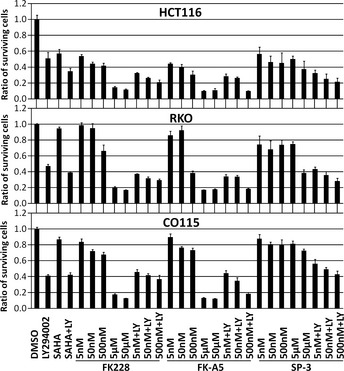
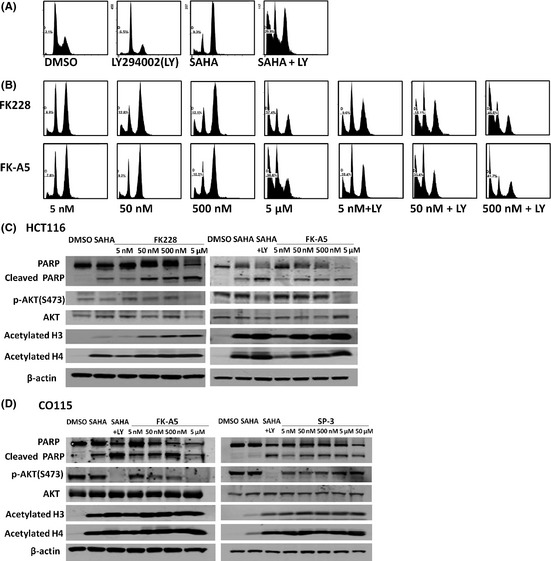
Activation of phosphatidylinositol 3-kinase (PI3K) signaling is involved in carcinogenesis and cancer progression. The PI3K inhibitors are considered candidate drugs for cancer treatment. Here, we describe a drug screening system for novel PI3K inhibitors using Saccharomyces cerevisiae strains with deleterious mutations in the ATP-binding cassette transporter genes, because wild-type S. cerevisiae uses drug efflux pumps for reducing intracellular drug concentrations. By screening the chemical library of the Screening Committee of Anticancer Drugs, we identified the histone deacetylase (HDAC) inhibitor romidepsin (FK228) and its novel analogs. In vitro PI3K activity assays confirmed that these compounds directly inhibit PI3K activity at μM-range concentrations. FK-A5 analog was the most potent inhibitor. Western blotting revealed that these compounds inhibit phosphorylation of protein kinase B and downstream signaling components. Molecular modeling of the PI3K-FK228 complex indicated that FK228 binds to the ATP-binding pocket of PI3K. At μM-range concentrations, FK228 and FK-A5 show potent cytotoxicity, inducing apoptosis even in HDAC inhibitor-resistant cells. Furthermore, HDAC/PI3K dual inhibition by FK228 and FK-A5 at μM-range concentrations potentiates the apoptosis induction, mimicking the effect of combining specific HDAC and PI3K inhibitors. In this study, we showed that FK228 and its analogs directly inhibit PI3K activity and induce apoptosis at μM-range concentrations, similar to HDAC/PI3K dual inhibition. In future, optimizing the potency of FK228 and its analogs against PI3K may contribute to the development of novel HDAC/PI3K dual inhibitors for cancer treatment.
© 2012 Japanese Cancer Association.
Figures
Similar articles
-
Biochemical, biological and structural properties of romidepsin (FK228) and its analogs as novel HDAC/PI3K dual inhibitors.Cancer Sci. 2015 Feb;106(2):208-15. doi: 10.1111/cas.12585. Epub 2015 Jan 28. Cancer Sci. 2015. PMID: 25492515 Free PMC article.
-
Antitumor activity and pharmacologic characterization of the depsipeptide analog as a novel histone deacetylase/ phosphatidylinositol 3-kinase dual inhibitor.Cancer Sci. 2017 Jul;108(7):1469-1475. doi: 10.1111/cas.13255. Epub 2017 May 19. Cancer Sci. 2017. PMID: 28406576 Free PMC article.
-
Romidepsin (FK228), A Histone Deacetylase Inhibitor and its Analogues in Cancer Chemotherapy.Curr Med Chem. 2021;28(7):1290-1303. doi: 10.2174/0929867327666200203113926. Curr Med Chem. 2021. PMID: 32013816 Review.
-
FK228 augmented temozolomide sensitivity in human glioma cells by blocking PI3K/AKT/mTOR signal pathways.Biomed Pharmacother. 2016 Dec;84:462-469. doi: 10.1016/j.biopha.2016.09.051. Epub 2016 Sep 28. Biomed Pharmacother. 2016. PMID: 27685789
-
FK228 (depsipeptide): a HDAC inhibitor with pleiotropic antitumor activities.Cancer Chemother Pharmacol. 2006 Nov;58(5):711-5. doi: 10.1007/s00280-005-0182-5. Epub 2006 Jan 25. Cancer Chemother Pharmacol. 2006. PMID: 16435156 Review.
Cited by
-
Dual Inhibitors Against Topoisomerases and Histone Deacetylases.J Cancer Prev. 2015 Jun;20(2):85-91. doi: 10.15430/JCP.2015.20.2.85. J Cancer Prev. 2015. PMID: 26151040 Free PMC article. Review.
-
The Role of the Unbinding Cycle on the Coordination Abilities of the Bi-Cyclopeptides toward Cu(II) Ions.Molecules. 2024 May 8;29(10):2197. doi: 10.3390/molecules29102197. Molecules. 2024. PMID: 38792059 Free PMC article.
-
Histone Deacetylase Inhibitors as Multitarget-Directed Epi-Drugs in Blocking PI3K Oncogenic Signaling: A Polypharmacology Approach.Int J Mol Sci. 2020 Nov 2;21(21):8198. doi: 10.3390/ijms21218198. Int J Mol Sci. 2020. PMID: 33147762 Free PMC article. Review.
-
Selenium protects against cadmium-induced kidney apoptosis in chickens by activating the PI3K/AKT/Bcl-2 signaling pathway.Environ Sci Pollut Res Int. 2017 Sep;24(25):20342-20353. doi: 10.1007/s11356-017-9422-6. Epub 2017 Jul 13. Environ Sci Pollut Res Int. 2017. PMID: 28707237
-
Mechanistic differences underlying HIV latency in the gut and blood contribute to differential responses to latency-reversing agents.AIDS. 2020 Nov 15;34(14):2013-2024. doi: 10.1097/QAD.0000000000002684. AIDS. 2020. PMID: 32910065 Free PMC article.
References
-
- Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu Rev Biochem 1998; 67: 481–507. - PubMed
-
- Cantley LC. The phosphoinositide 3‐kinase pathway. Science 2002; 296: 1655–7. - PubMed
-
- Samuels Y, Wang Z, Bardelli A et al High frequency of mutations of the PIK3CA gene in human cancers. Science 2004; 304: 554. - PubMed
-
- Ikenoue T, Kanai F, Hikiba Y et al Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res 2005; 65: 4562–7. - PubMed
-
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5‐trisphosphate. J Biol Chem 1998; 273: 13375–8. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
