

Rapid phylogenetic and functional classification of short genomic fragments with signature peptides
- PMID: 22925230
- PMCID: PMC3772700
- DOI: 10.1186/1756-0500-5-460
Rapid phylogenetic and functional classification of short genomic fragments with signature peptides
Abstract
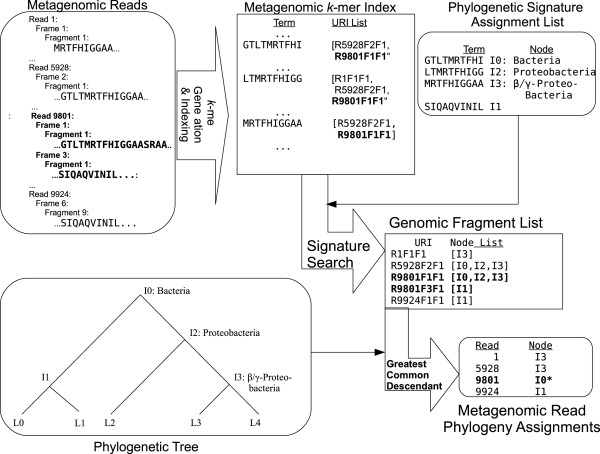
Background: Classification is difficult for shotgun metagenomics data from environments such as soils, where the diversity of sequences is high and where reference sequences from close relatives may not exist. Approaches based on sequence-similarity scores must deal with the confounding effects that inheritance and functional pressures exert on the relation between scores and phylogenetic distance, while approaches based on sequence alignment and tree-building are typically limited to a small fraction of gene families. We describe an approach based on finding one or more exact matches between a read and a precomputed set of peptide 10-mers.
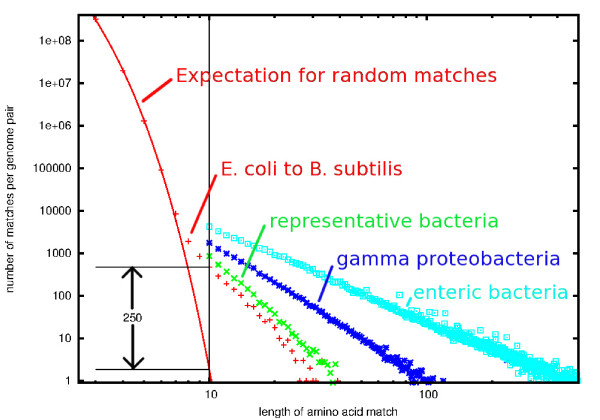
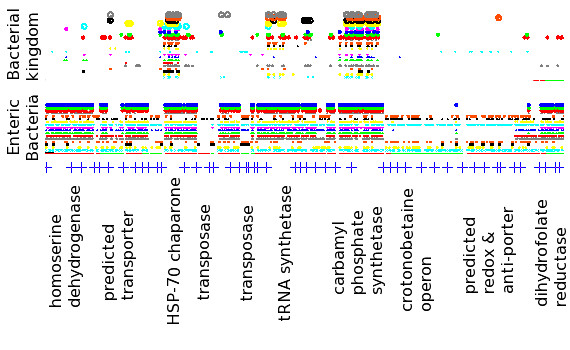
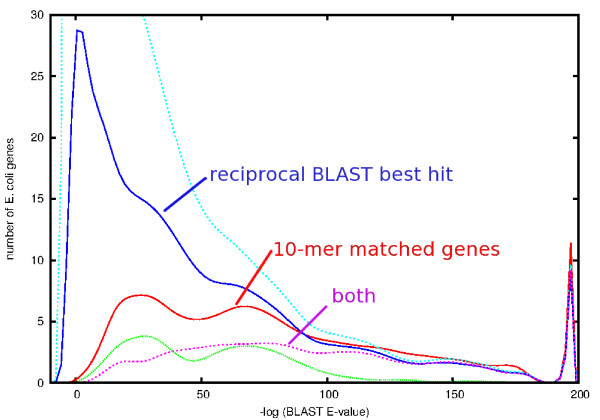
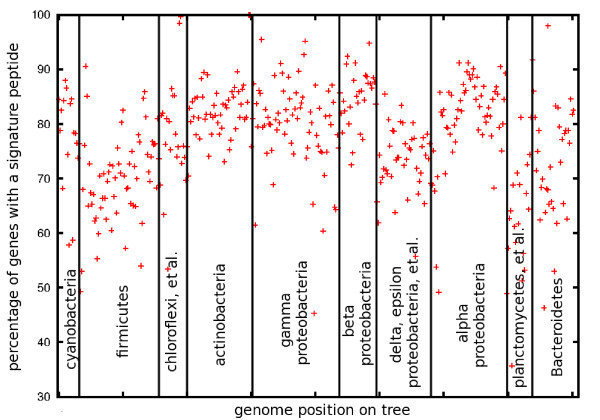
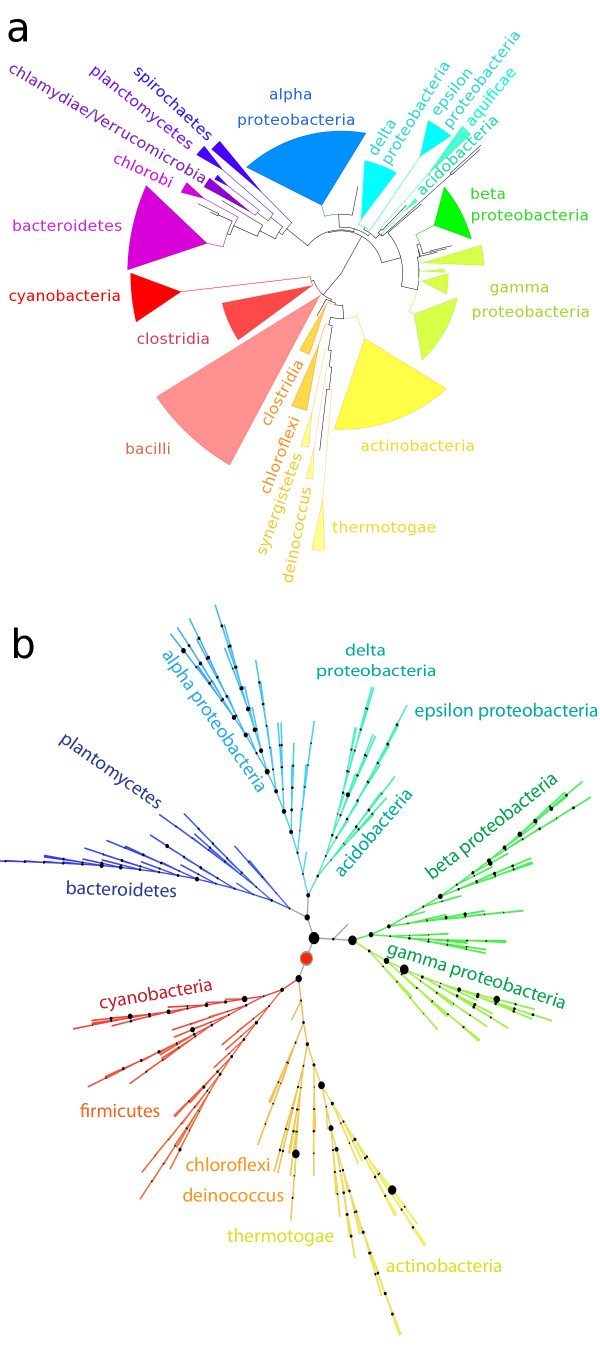
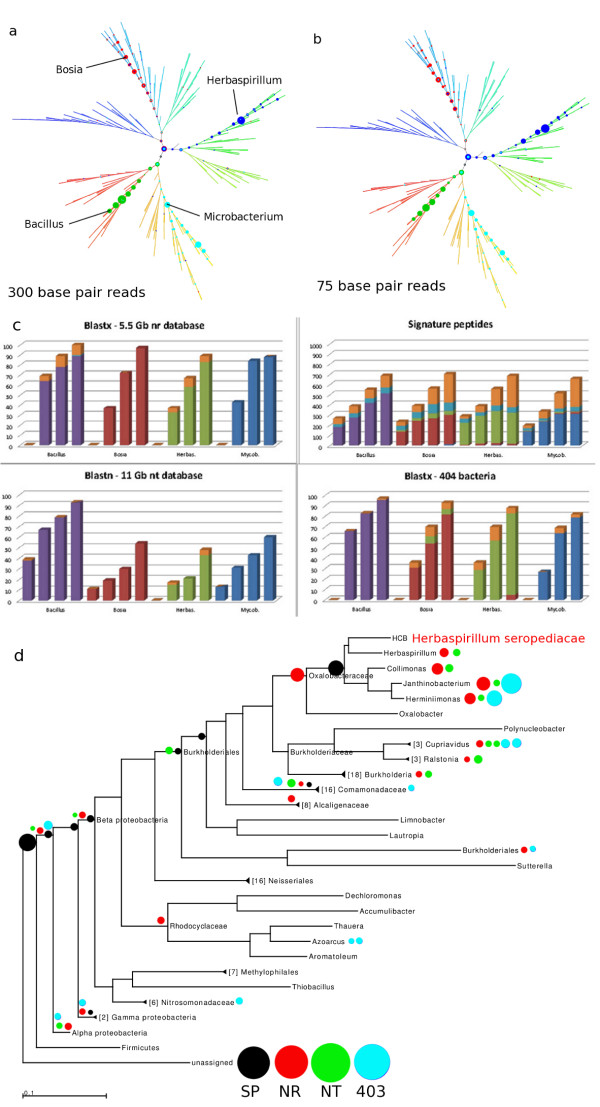
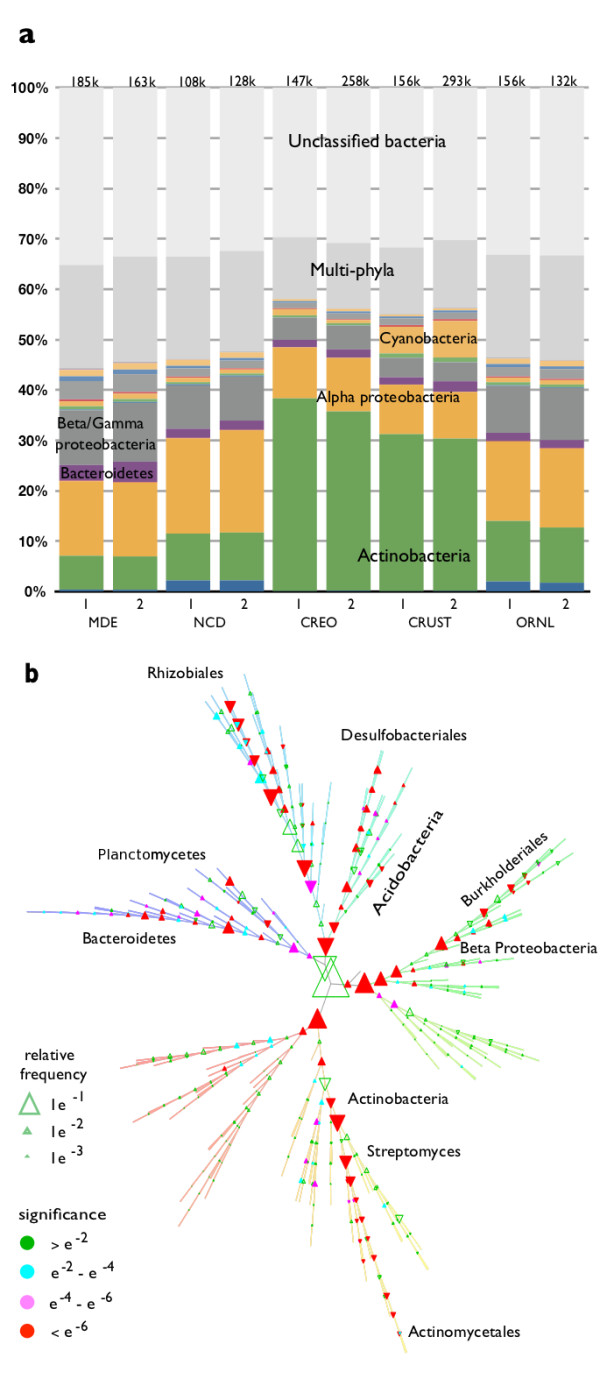
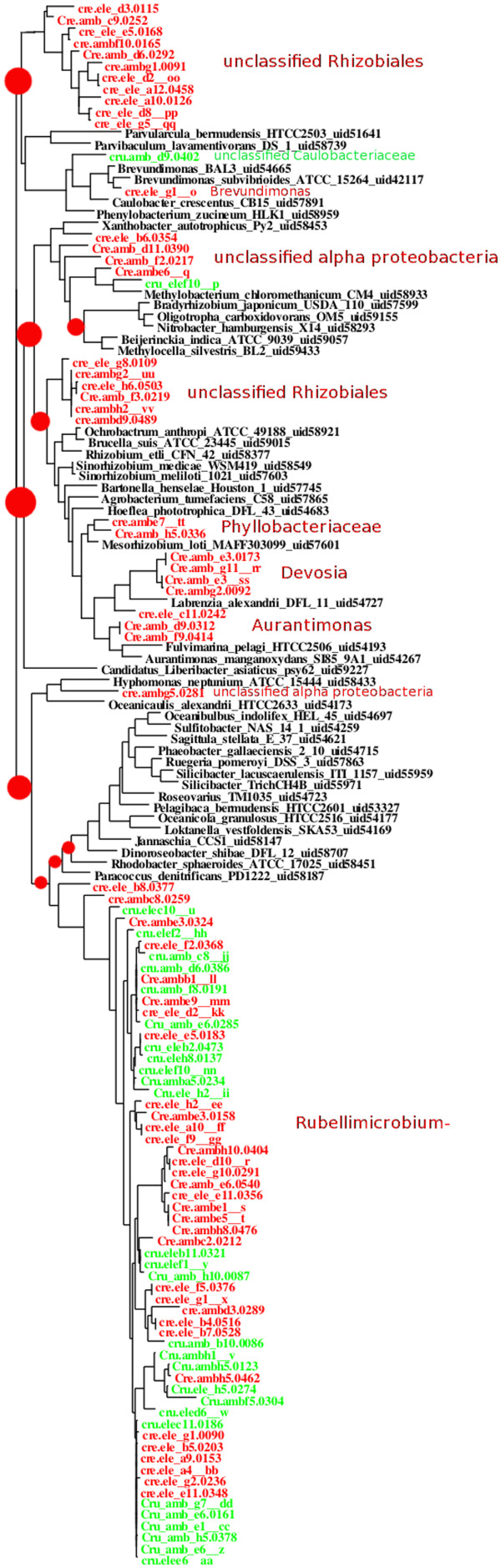
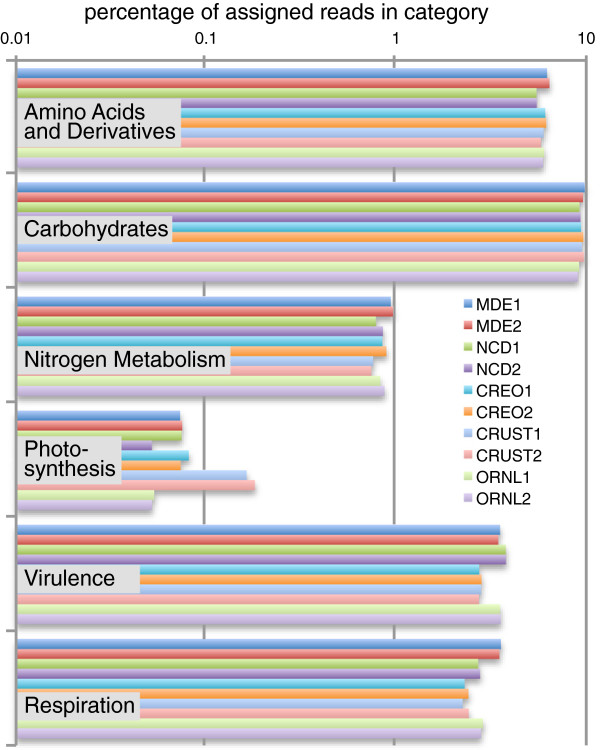
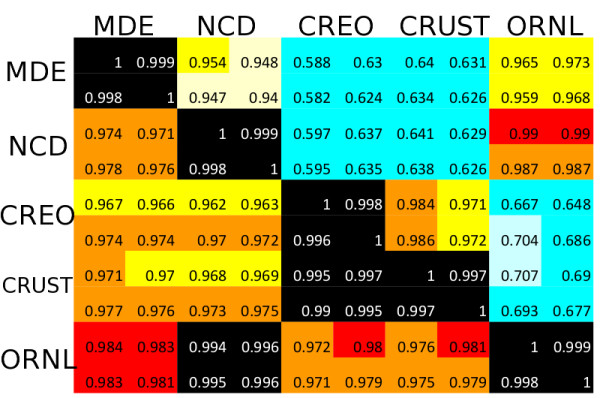
Results: At even the largest phylogenetic distances, thousands of 10-mer peptide exact matches can be found between pairs of bacterial genomes. Genes that share one or more peptide 10-mers typically have high reciprocal BLAST scores. Among a set of 403 representative bacterial genomes, some 20 million 10-mer peptides were found to be shared. We assign each of these peptides as a signature of a particular node in a phylogenetic reference tree based on the RNA polymerase genes. We classify the phylogeny of a genomic fragment (e.g., read) at the most specific node on the reference tree that is consistent with the phylogeny of observed signature peptides it contains. Using both synthetic data from four newly-sequenced soil-bacterium genomes and ten real soil metagenomics data sets, we demonstrate a sensitivity and specificity comparable to that of the MEGAN metagenomics analysis package using BLASTX against the NR database. Phylogenetic and functional similarity metrics applied to real metagenomics data indicates a signal-to-noise ratio of approximately 400 for distinguishing among environments. Our method assigns ~6.6 Gbp/hr on a single CPU, compared with 25 kbp/hr for methods based on BLASTX against the NR database.
Conclusions: Classification by exact matching against a precomputed list of signature peptides provides comparable results to existing techniques for reads longer than about 300 bp and does not degrade severely with shorter reads. Orders of magnitude faster than existing methods, the approach is suitable now for inclusion in analysis pipelines and appears to be extensible in several different directions.
Figures

Similar articles
-
Beyond classification: gene-family phylogenies from shotgun metagenomic reads enable accurate community analysis.BMC Genomics. 2013 Jun 22;14:419. doi: 10.1186/1471-2164-14-419. BMC Genomics. 2013. PMID: 23799973 Free PMC article.
-
Short clones or long clones? A simulation study on the use of paired reads in metagenomics.BMC Bioinformatics. 2010 Jan 18;11 Suppl 1(Suppl 1):S12. doi: 10.1186/1471-2105-11-S1-S12. BMC Bioinformatics. 2010. PMID: 20122183 Free PMC article.
-
RAIphy: phylogenetic classification of metagenomics samples using iterative refinement of relative abundance index profiles.BMC Bioinformatics. 2011 Jan 31;12:41. doi: 10.1186/1471-2105-12-41. BMC Bioinformatics. 2011. PMID: 21281493 Free PMC article.
-
ReprDB and panDB: minimalist databases with maximal microbial representation.Microbiome. 2018 Jan 18;6(1):15. doi: 10.1186/s40168-018-0399-2. Microbiome. 2018. PMID: 29347966 Free PMC article.
-
Reference databases for taxonomic assignment in metagenomics.Brief Bioinform. 2012 Nov;13(6):682-95. doi: 10.1093/bib/bbs036. Epub 2012 Jul 10. Brief Bioinform. 2012. PMID: 22786784 Review.
Cited by
-
Scalable metagenomic taxonomy classification using a reference genome database.Bioinformatics. 2013 Sep 15;29(18):2253-60. doi: 10.1093/bioinformatics/btt389. Epub 2013 Jul 4. Bioinformatics. 2013. PMID: 23828782 Free PMC article.
-
Evaluation of shotgun metagenomics sequence classification methods using in silico and in vitro simulated communities.BMC Bioinformatics. 2015 Nov 4;16:363. doi: 10.1186/s12859-015-0788-5. BMC Bioinformatics. 2015. PMID: 26537885 Free PMC article.
-
SUPER-FOCUS: a tool for agile functional analysis of shotgun metagenomic data.Bioinformatics. 2016 Feb 1;32(3):354-61. doi: 10.1093/bioinformatics/btv584. Epub 2015 Oct 9. Bioinformatics. 2016. PMID: 26454280 Free PMC article.
-
Environmental genes and genomes: understanding the differences and challenges in the approaches and software for their analyses.Brief Bioinform. 2015 Sep;16(5):745-58. doi: 10.1093/bib/bbv001. Epub 2015 Feb 11. Brief Bioinform. 2015. PMID: 25673291 Free PMC article.
-
California condor microbiomes: Bacterial variety and functional properties in captive-bred individuals.PLoS One. 2019 Dec 11;14(12):e0225858. doi: 10.1371/journal.pone.0225858. eCollection 2019. PLoS One. 2019. PMID: 31825977 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
