

CCL2/CCR2 chemokine signaling coordinates survival and motility of breast cancer cells through Smad3 protein- and p42/44 mitogen-activated protein kinase (MAPK)-dependent mechanisms
- PMID: 22927430
- PMCID: PMC3476325
- DOI: 10.1074/jbc.M112.365999
CCL2/CCR2 chemokine signaling coordinates survival and motility of breast cancer cells through Smad3 protein- and p42/44 mitogen-activated protein kinase (MAPK)-dependent mechanisms
Abstract
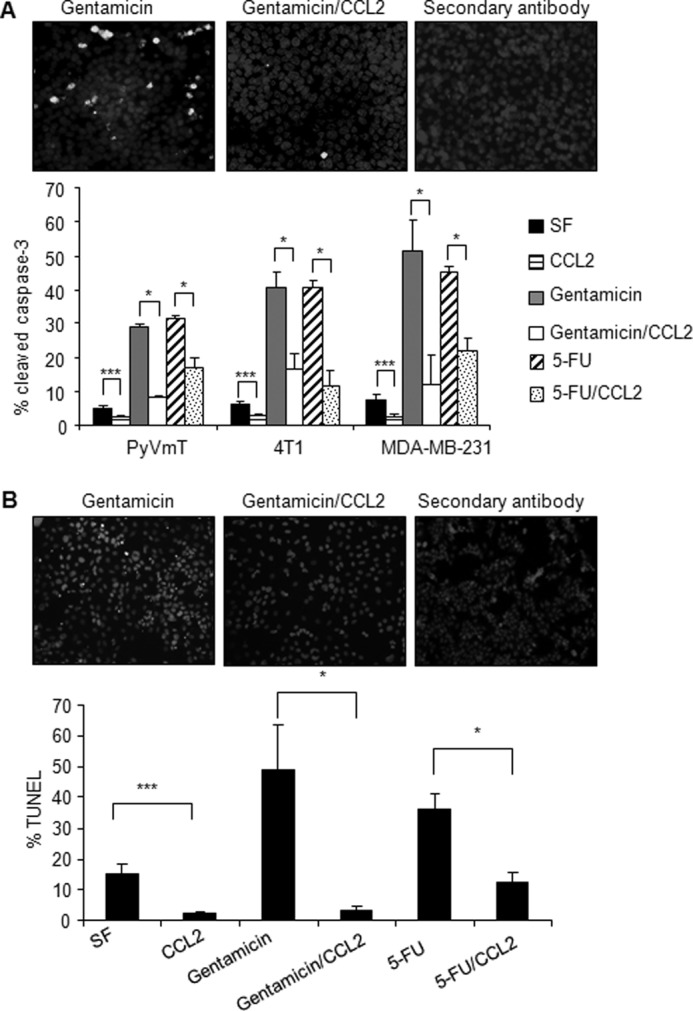
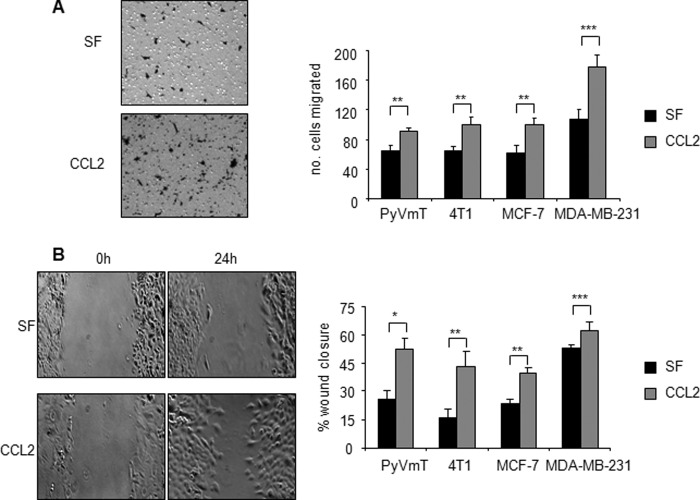
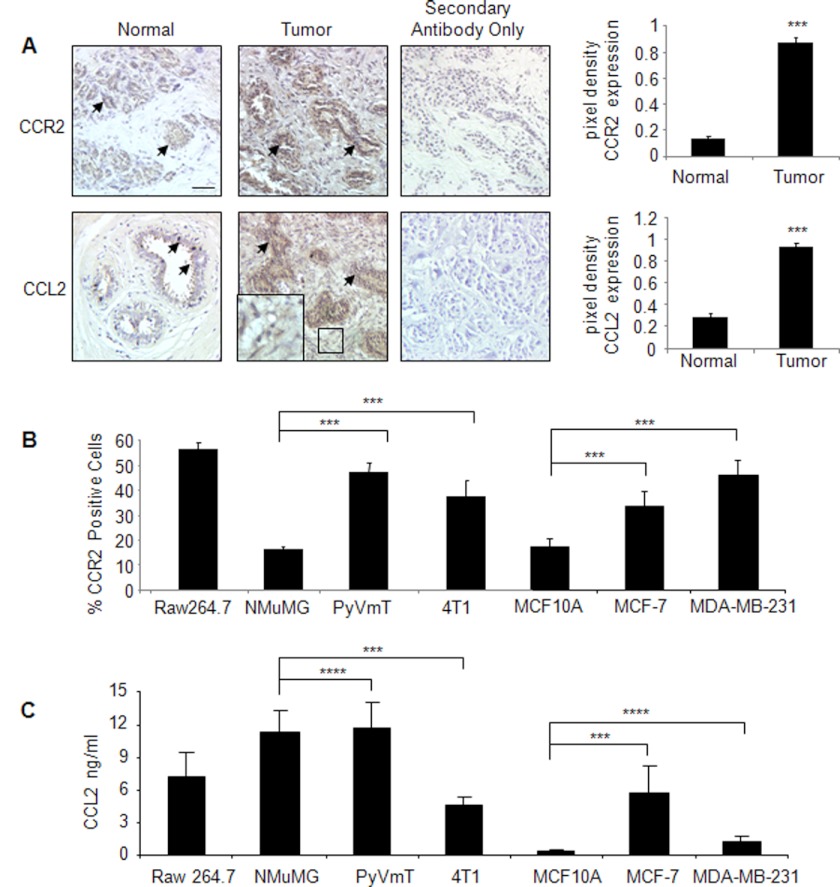
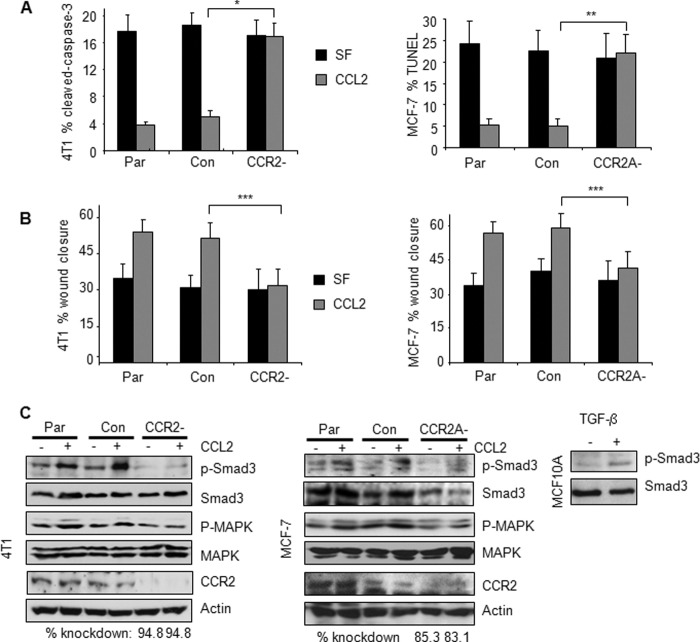
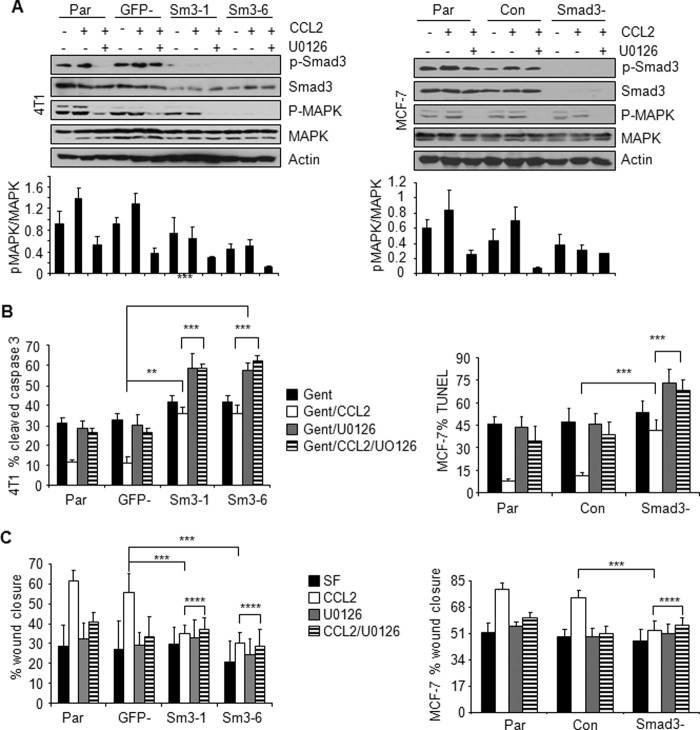
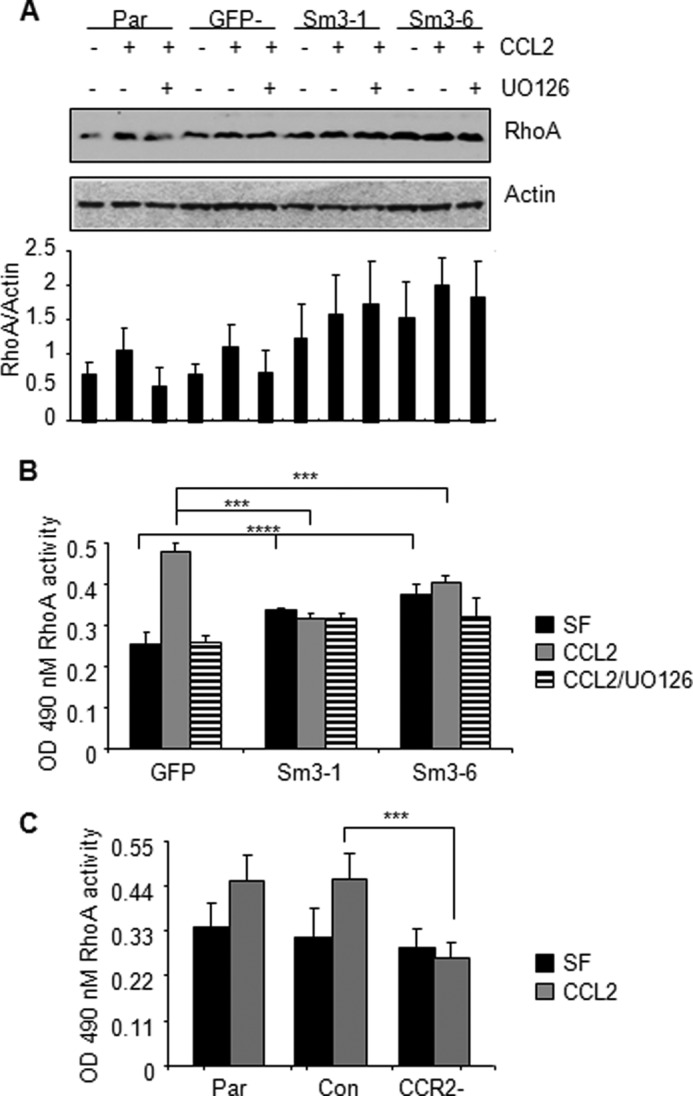
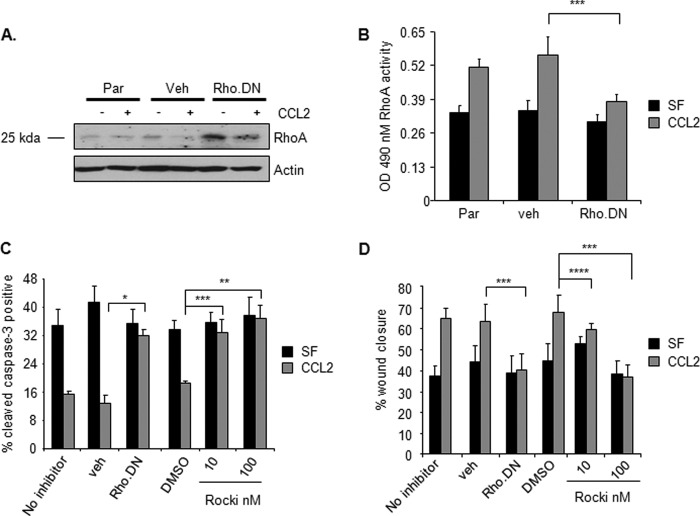
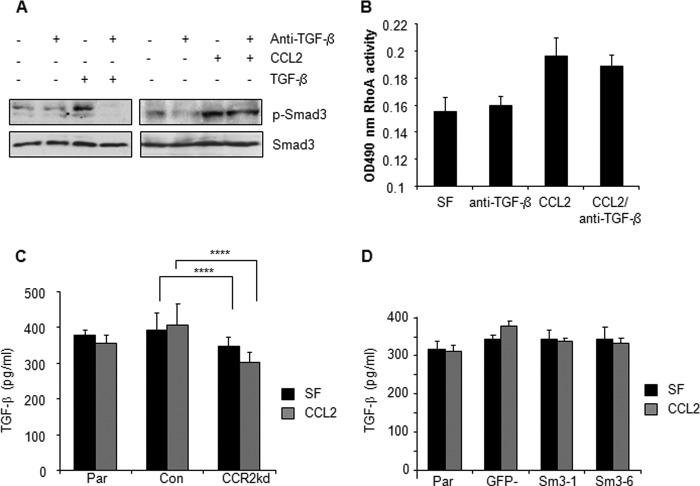
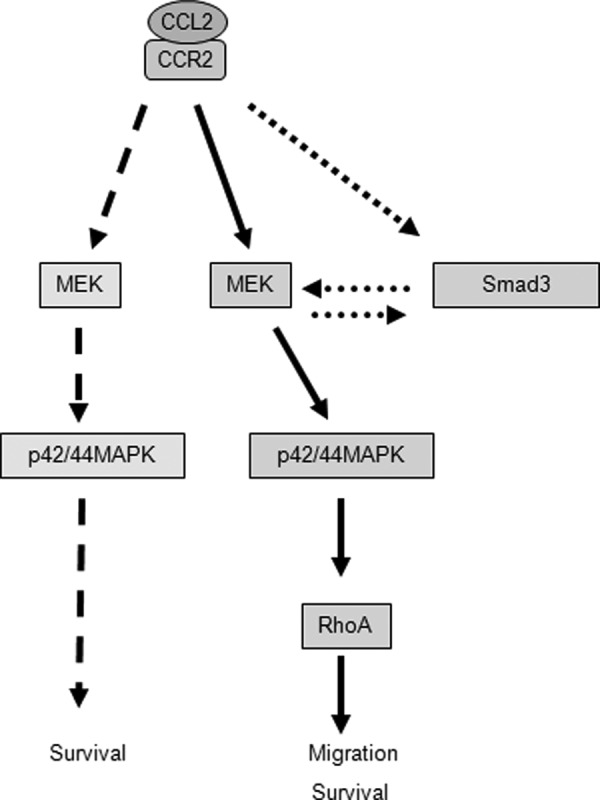
Increased cell motility and survival are important hallmarks of metastatic tumor cells. However, the mechanisms that regulate the interplay between these cellular processes remain poorly understood. In these studies, we demonstrate that CCL2, a chemokine well known for regulating immune cell migration, plays an important role in signaling to breast cancer cells. We report that in a panel of mouse and human breast cancer cell lines CCL2 enhanced cell migration and survival associated with increased phosphorylation of Smad3 and p42/44MAPK proteins. The G protein-coupled receptor CCR2 was found to be elevated in breast cancers, correlating with CCL2 expression. RNA interference of CCR2 expression in breast cancer cells significantly inhibited CCL2-induced migration, survival, and phosphorylation of Smad3 and p42/44MAPK proteins. Disruption of Smad3 expression in mammary carcinoma cells blocked CCL2-induced cell survival and migration and partially reduced p42/44MAPK phosphorylation. Ablation of MAPK phosphorylation in Smad3-deficient cells with the MEK inhibitor U0126 further reduced cell survival but not migration. These data indicate that Smad3 signaling through MEK-p42/44MAPK regulates CCL2-induced cell motility and survival, whereas CCL2 induction of MEK-p42/44MAPK signaling independent of Smad3 functions as an alternative mechanism for cell survival. Furthermore, we show that CCL2-induced Smad3 signaling through MEK-p42/44MAPK regulates expression and activity of Rho GTPase to mediate CCL2-induced breast cancer cell motility and survival. With these studies, we characterize an important role for CCL2/CCR2 chemokine signaling in regulating the intrinsic relationships between breast cancer cell motility and survival with implications on the metastatic process.
Figures
References
-
- Gillitzer R., Goebeler M. (2001) Chemokines in cutaneous wound healing. J. Leukoc. Biol. 69, 513–521 - PubMed
-
- Laing K. J., Secombes C. J. (2004) Chemokines. Dev. Comp. Immunol. 28, 443–460 - PubMed
-
- Lentsch A. B. (2002) The Duffy antigen/receptor for chemokines (DARC) and prostate cancer. A role as clear as black and white? FASEB J. 16, 1093–1095 - PubMed
-
- Ernst C. A., Zhang Y. J., Hancock P. R., Rutledge B. J., Corless C. L., Rollins B. J. (1994) Biochemical and biologic characterization of murine monocyte chemoattractant protein-1. Identification of two functional domains. J. Immunol. 152, 3541–3549 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
