

Genometa--a fast and accurate classifier for short metagenomic shotgun reads
- PMID: 22927906
- PMCID: PMC3424124
- DOI: 10.1371/journal.pone.0041224
Genometa--a fast and accurate classifier for short metagenomic shotgun reads
Abstract
Metagenomic studies use high-throughput sequence data to investigate microbial communities in situ. However, considerable challenges remain in the analysis of these data, particularly with regard to speed and reliable analysis of microbial species as opposed to higher level taxa such as phyla. We here present Genometa, a computationally undemanding graphical user interface program that enables identification of bacterial species and gene content from datasets generated by inexpensive high-throughput short read sequencing technologies. Our approach was first verified on two simulated metagenomic short read datasets, detecting 100% and 94% of the bacterial species included with few false positives or false negatives. Subsequent comparative benchmarking analysis against three popular metagenomic algorithms on an Illumina human gut dataset revealed Genometa to attribute the most reads to bacteria at species level (i.e. including all strains of that species) and demonstrate similar or better accuracy than the other programs. Lastly, speed was demonstrated to be many times that of BLAST due to the use of modern short read aligners. Our method is highly accurate if bacteria in the sample are represented by genomes in the reference sequence but cannot find species absent from the reference. This method is one of the most user-friendly and resource efficient approaches and is thus feasible for rapidly analysing millions of short reads on a personal computer.
Availability: The Genometa program, a step by step tutorial and Java source code are freely available from http://genomics1.mh-hannover.de/genometa/ and on http://code.google.com/p/genometa/. This program has been tested on Ubuntu Linux and Windows XP/7.
Conflict of interest statement
Figures
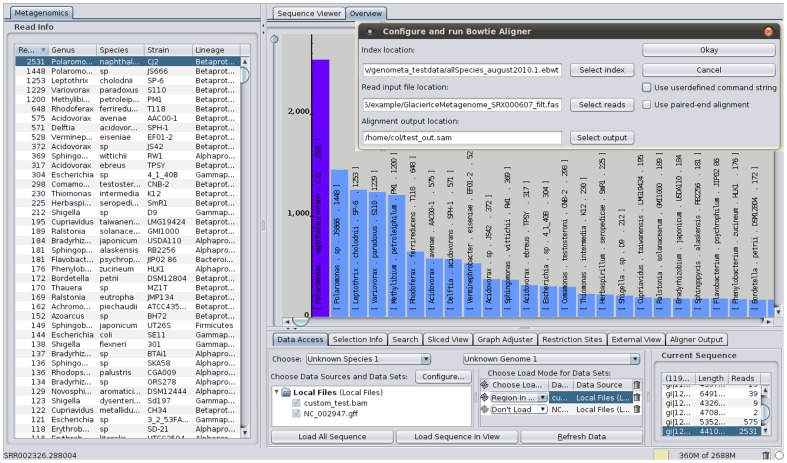
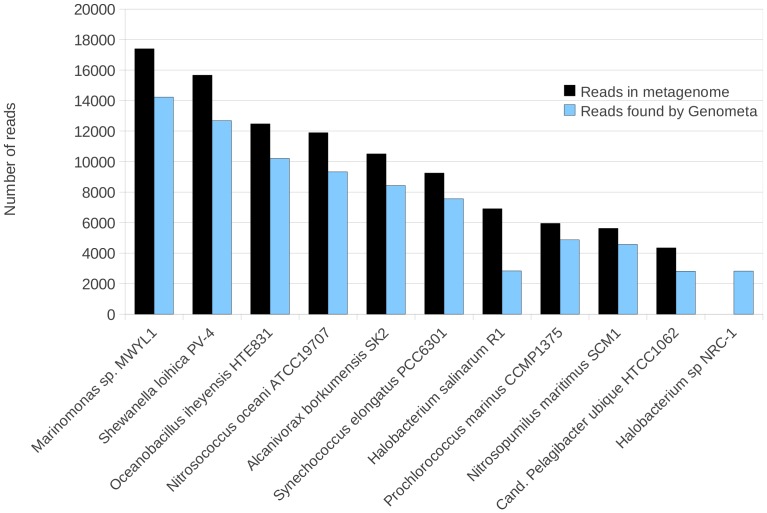
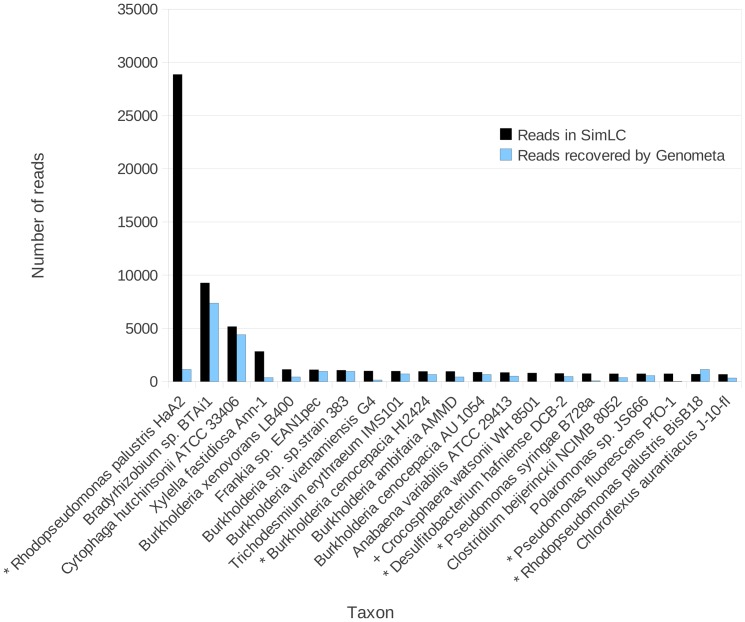
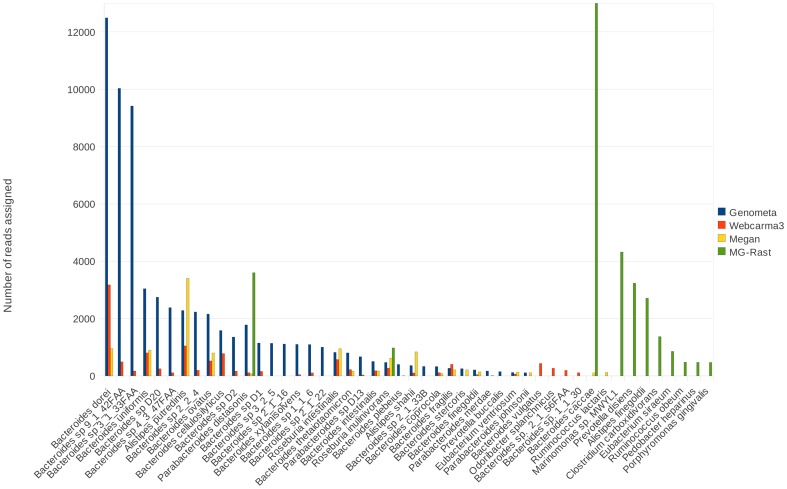
References
-
- Metzker ML (2010) Sequencing technologies - the next generation. Nat Rev Genet 11: 31–46. - PubMed
-
- Coetzee B, Freeborough M-J, Maree HJ, Celton JM, Rees DJG, et al. (2010) Deep sequencing analysis of viruses infecting grapevines: Virome of a vineyard. Virology 400: 157–163. - PubMed
-
- Hess M, Sczyrba A, Egan R, Kim TW, Chokhawala H, et al. (2011) Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 331: 463–467. - PubMed
-
- Shah N, Tang H, Doak TG, Ye Y (2011) Comparing bacterial communities inferred from 16s rRNA gene sequencing and shotgun metagenomics. Pac Symp Biocomput 165–176. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
