

Repair of diverse diabetic defects of β-cells in man and mouse by pharmacological glucokinase activation
- PMID: 22928571
- PMCID: PMC4433321
- DOI: 10.1111/j.1463-1326.2012.01652.x
Repair of diverse diabetic defects of β-cells in man and mouse by pharmacological glucokinase activation
Abstract
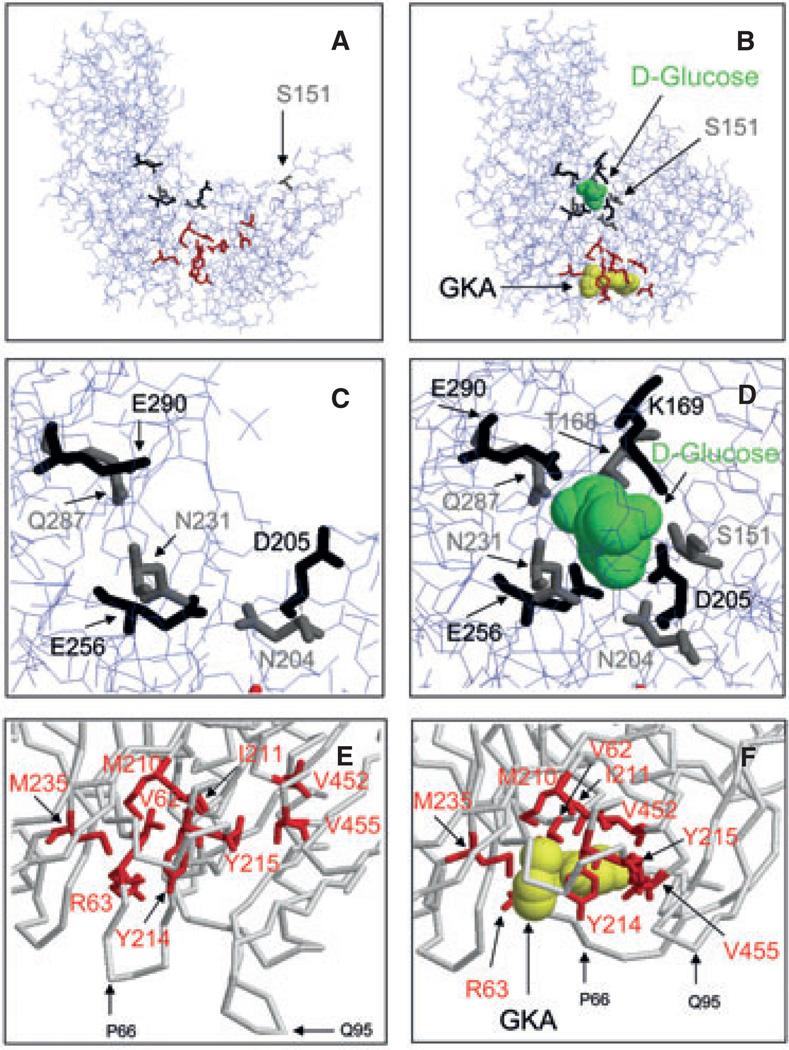
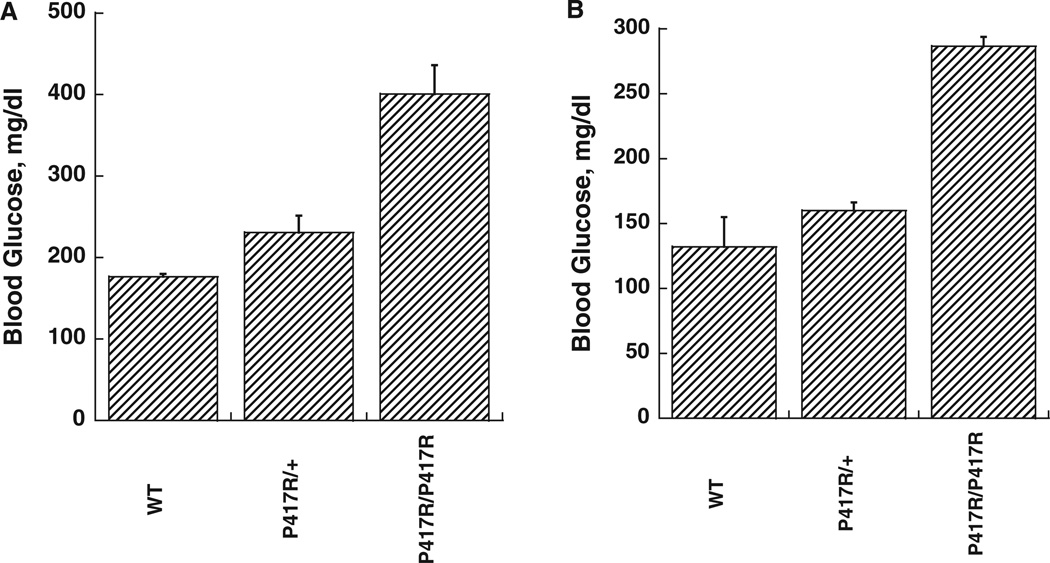
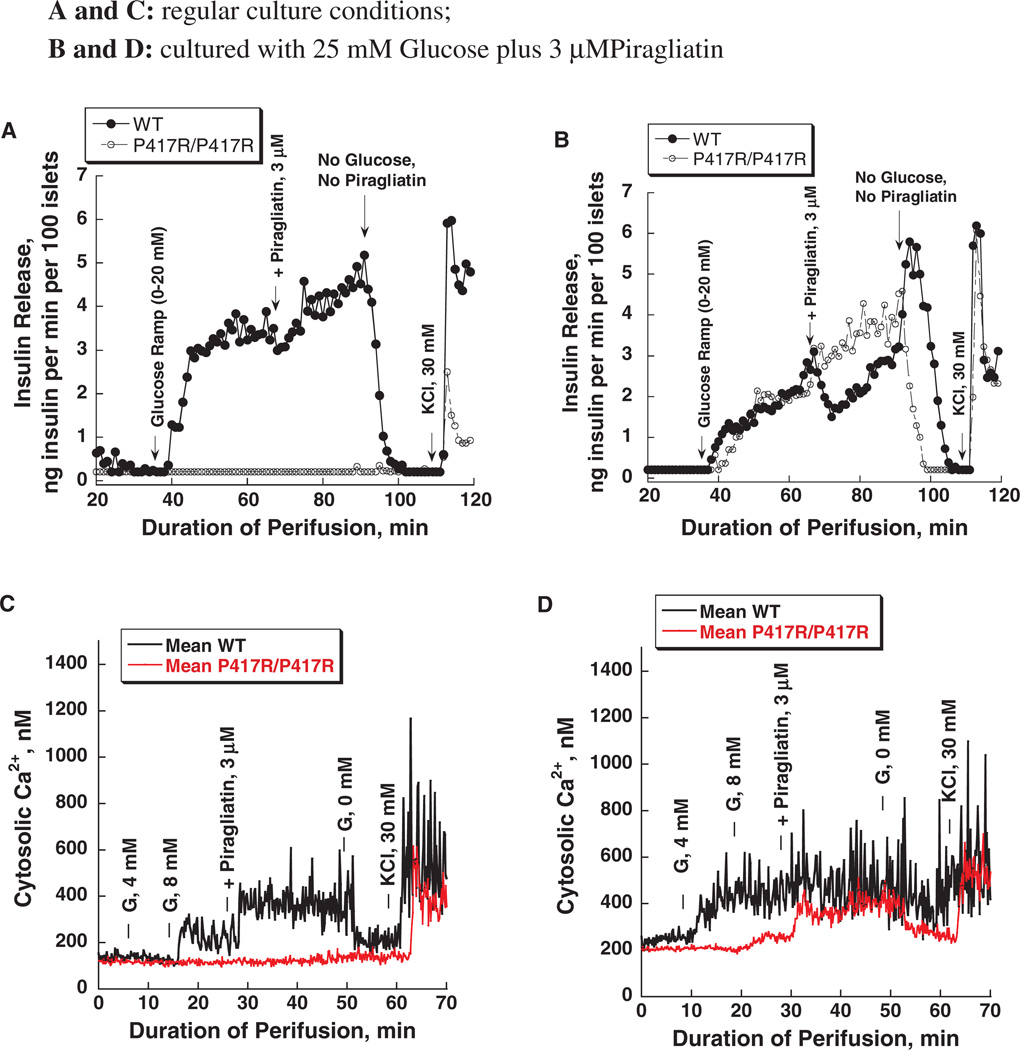
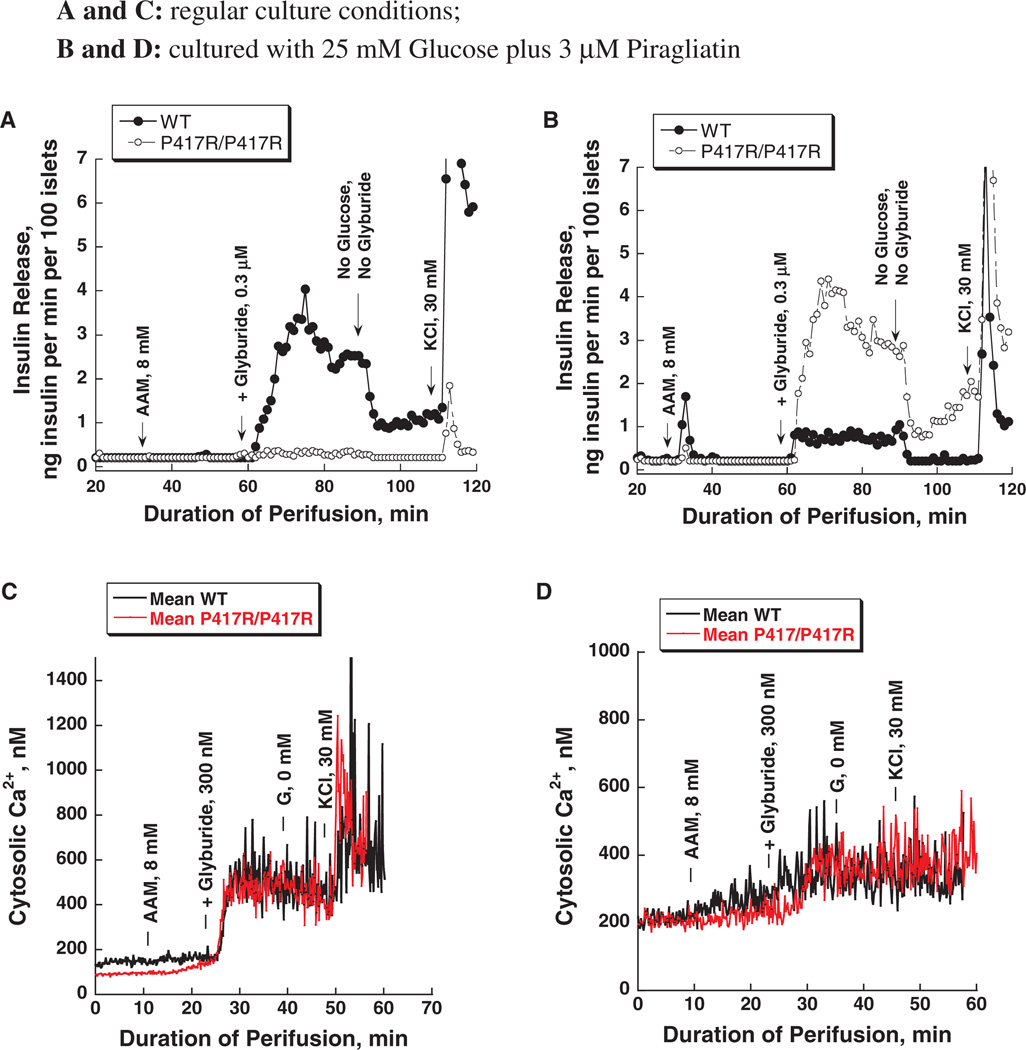
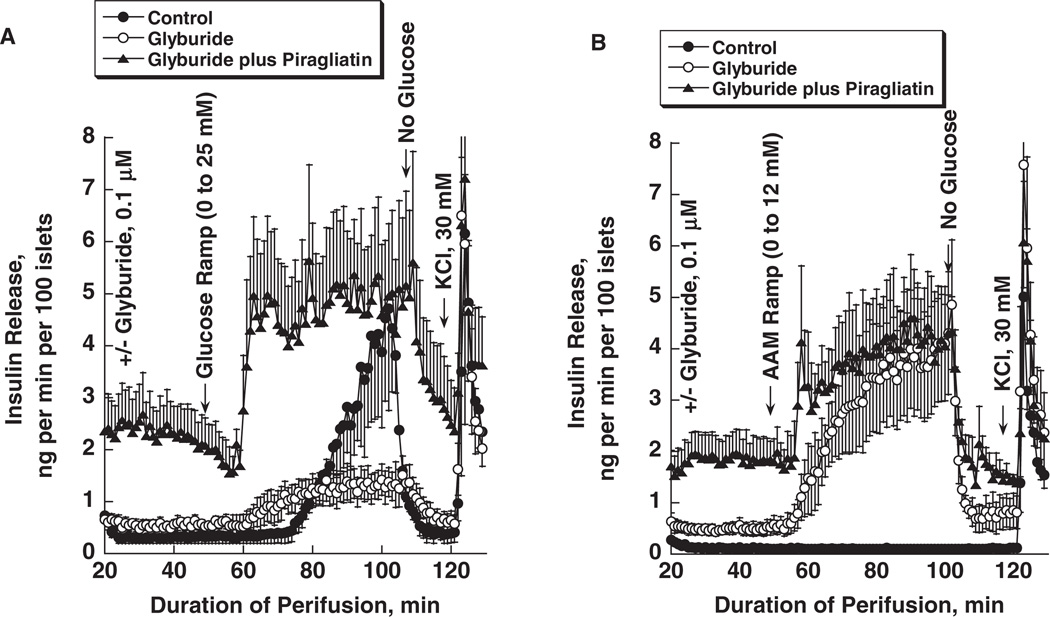
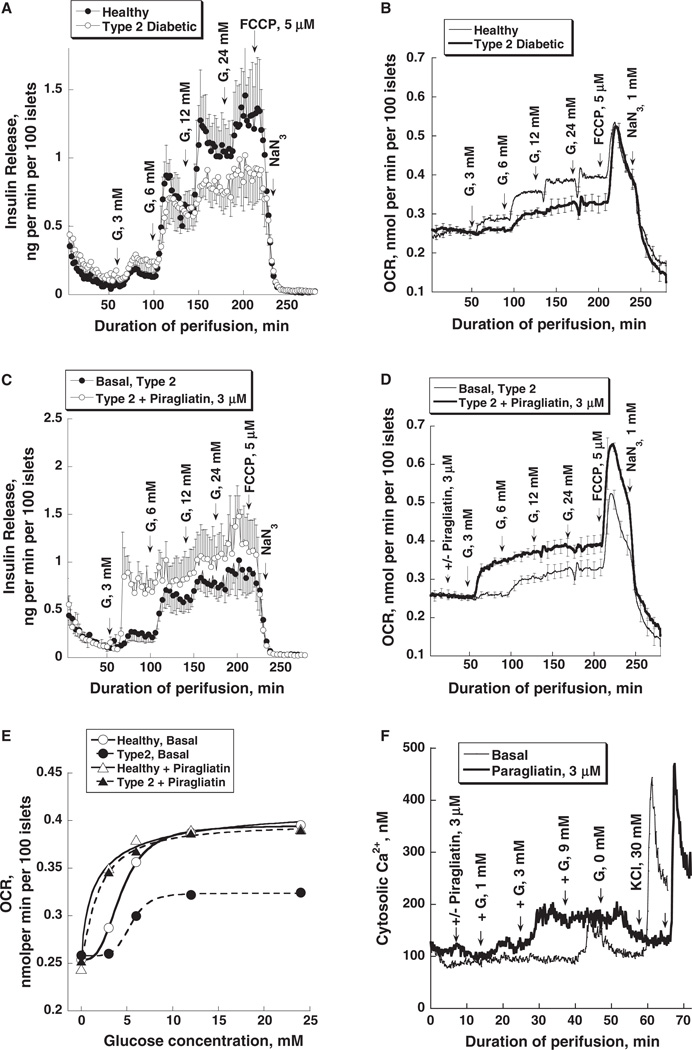
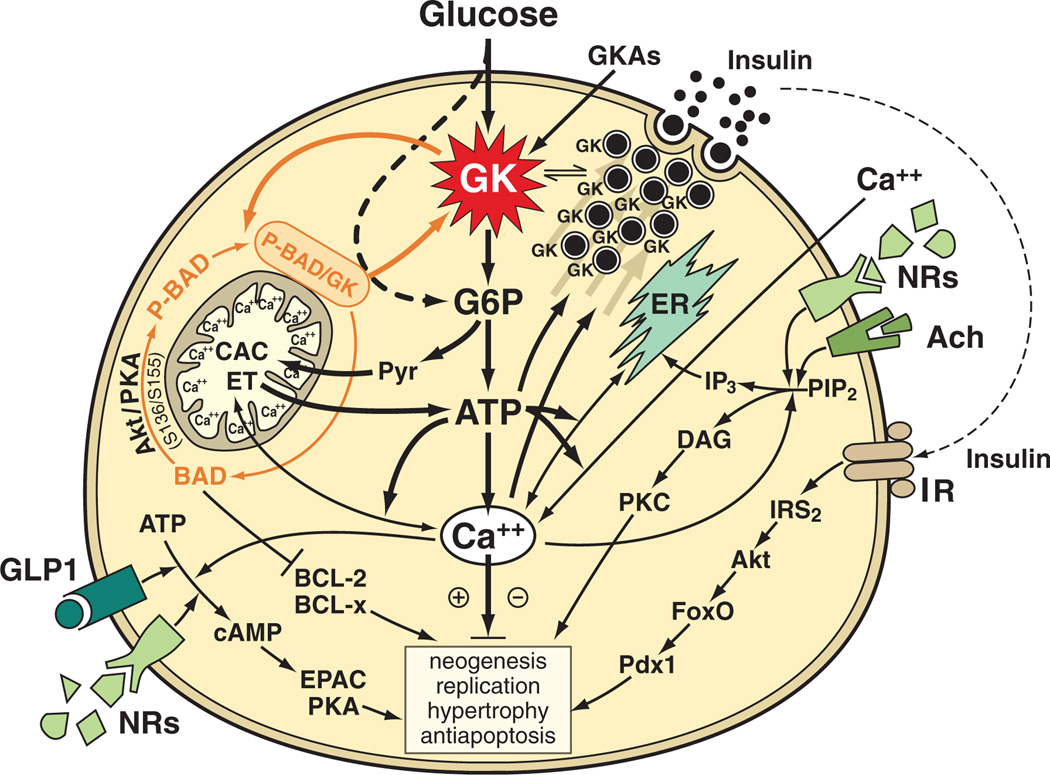
Glucokinase activators (GKAs) are being developed and clinically tested for potential antidiabetic therapy. The potential benefits and limitations of this approach continue to be intensively debated. To contribute to the understanding of experimental pharmacology and therapeutics of GKAs, we have tested the efficacy of one of these agents (Piragliatin) in isolated islets from humans with type 2 diabetes mellitus (T2DM), from mice with glucokinase (GK) mutations induced by ethyl-nitroso-urea (ENU) as models of Maturity Onset Diabetes of the Young linked to GK and Permanent Neonatal Diabetes Mellitus linked to GK (PNDM-GK) and finally of islets rendered glucose insensitive by treatment with the sulphonyl urea compound glyburide in organ culture. We found that the GKA repaired the defect in all three instances as manifest in increased glucose-induced insulin release and elevated intracellular calcium responses. The results show the remarkable fact that acute pharmacological activation of GK reverses secretion defects of β-cells caused by molecular mechanism that differ vastly in nature, including the little understood multifactorial lesion of β-cells in T2DM of man, the complex GK mutations in mice resembling GK disease and acute sulphonylurea failure of mouse β-cells in tissue culture. The implications of these results are to be discussed on the theoretical basis underpinning the strategy of developing these drugs and in light of recent results of clinical trials with GKAs that failed for little understood reasons.
© 2012 Blackwell Publishing Ltd.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Chronic glucokinase activator treatment at clinically translatable exposures gives durable glucose lowering in two animal models of type 2 diabetes.Br J Pharmacol. 2014 Apr;171(7):1642-54. doi: 10.1111/bph.12504. Br J Pharmacol. 2014. PMID: 24772484 Free PMC article.
-
Recent clinical advances of glucokinase activators in the treatment of diabetes mellitus type 2.Pharmazie. 2020 Jun 1;75(6):230-235. doi: 10.1691/ph.2020.0409. Pharmazie. 2020. PMID: 32539915 Review.
-
Novel, highly potent systemic glucokinase activators for the treatment of Type 2 Diabetes Mellitus.Bioorg Med Chem Lett. 2017 May 1;27(9):2069-2073. doi: 10.1016/j.bmcl.2016.10.085. Epub 2016 Oct 31. Bioorg Med Chem Lett. 2017. PMID: 28284804
-
Allosteric activators of glucokinase: potential role in diabetes therapy.Science. 2003 Jul 18;301(5631):370-3. doi: 10.1126/science.1084073. Science. 2003. PMID: 12869762
-
Recent updates on glucokinase activators for the treatment of type 2 diabetes mellitus.Mini Rev Med Chem. 2014;14(7):585-602. doi: 10.2174/1389557514666140722082713. Mini Rev Med Chem. 2014. PMID: 25052034 Review.
Cited by
-
Lipotoxicity in a Vicious Cycle of Pancreatic Beta Cell Exhaustion.Biomedicines. 2022 Jul 7;10(7):1627. doi: 10.3390/biomedicines10071627. Biomedicines. 2022. PMID: 35884932 Free PMC article.
-
Safety, tolerability, pharmacokinetics, and pharmacodynamics of novel glucokinase activator HMS5552: results from a first-in-human single ascending dose study.Drug Des Devel Ther. 2016 May 9;10:1619-26. doi: 10.2147/DDDT.S105021. eCollection 2016. Drug Des Devel Ther. 2016. PMID: 27274195 Free PMC article.
-
Control of insulin secretion by cytochrome C and calcium signaling in islets with impaired metabolism.J Biol Chem. 2014 Jul 4;289(27):19110-9. doi: 10.1074/jbc.M114.556050. Epub 2014 May 19. J Biol Chem. 2014. PMID: 24841202 Free PMC article.
-
Efficacy and safety of dorzagliatin for type 2 diabetes mellitus: A meta-analysis and trial sequential analysis.Front Cardiovasc Med. 2022 Nov 23;9:1041044. doi: 10.3389/fcvm.2022.1041044. eCollection 2022. Front Cardiovasc Med. 2022. PMID: 36505359 Free PMC article.
-
Normal glucose metabolism in carnivores overlaps with diabetes pathology in non-carnivores.Front Endocrinol (Lausanne). 2013 Dec 3;4:188. doi: 10.3389/fendo.2013.00188. Front Endocrinol (Lausanne). 2013. PMID: 24348462 Free PMC article. Review.
References
-
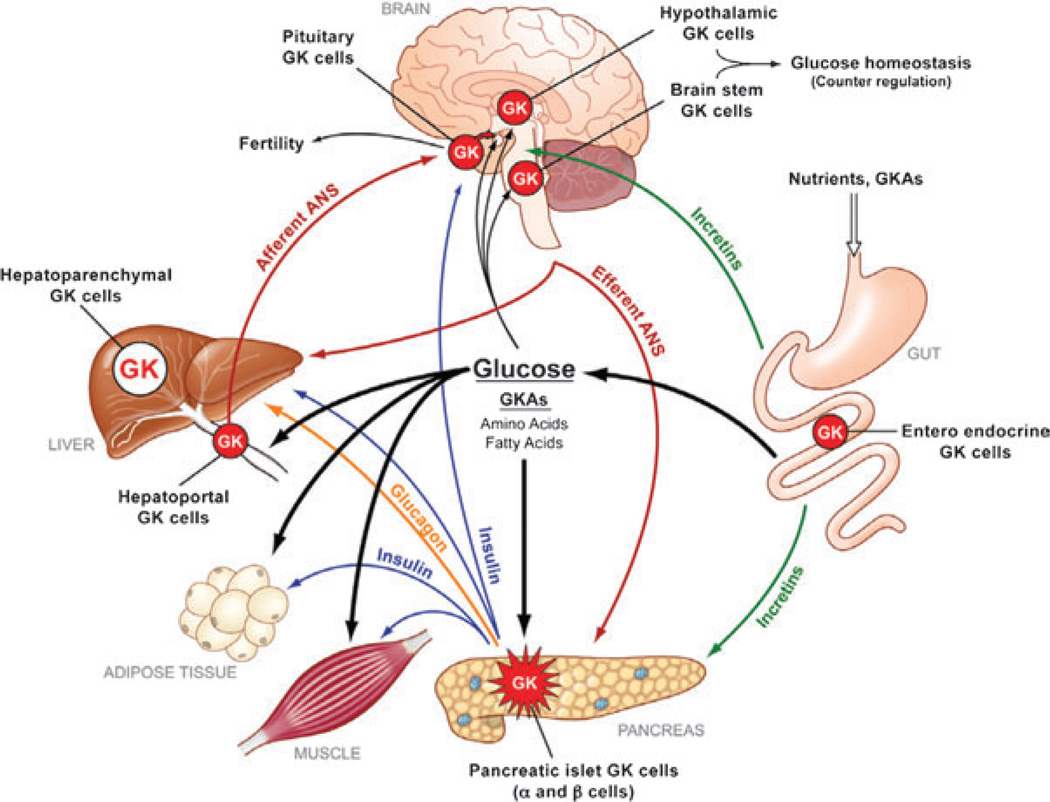
- Matschinsky FM, Magnuson MA, Zelent D, et al. The network of glucokinase-expressing cells in glucose homeostasis and the potential of glucokinase activators for diabetes therapy. Diabetes. 2006;55:1–12. - PubMed
-
- Cuesta-Munoz AL, Huopio H, Otonkoski T, et al. Severe persistent hyperinsulinemic hypoglycemia due to a de novo glucokinase mutation. Diabetes. 2004;53:2164–2168. - PubMed
-
- Njolstad PR, Sovik O, Cuesta-Munoz A, Bjorkhaug L, et al. Neonatal diabetes mellitus due to complete glucokinase deficiency. N Engl J Med. 2001;344:1588–1592. - PubMed
-
- Njolstad PR, Sagen JV, Bjorkhaug L, et al. Permanent neonatal diabetes caused by glucokinase deficiency: inborn error of the glucose-insulin signaling pathway. Diabetes. 2003;52:2854–60. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
