

Therapeutic impact of follistatin-like 1 on myocardial ischemic injury in preclinical models
- PMID: 22929303
- PMCID: PMC3548325
- DOI: 10.1161/CIRCULATIONAHA.112.115089
Therapeutic impact of follistatin-like 1 on myocardial ischemic injury in preclinical models
Abstract
Background: Acute coronary syndrome is a leading cause of death in developed countries. Follistatin-like 1 (FSTL1) is a myocyte-derived secreted protein that is upregulated in the heart in response to ischemic insult. Here, we investigated the therapeutic impact of FSTL1 on acute cardiac injury in small and large preclinical animal models of ischemia/reperfusion and dissected its molecular mechanism.
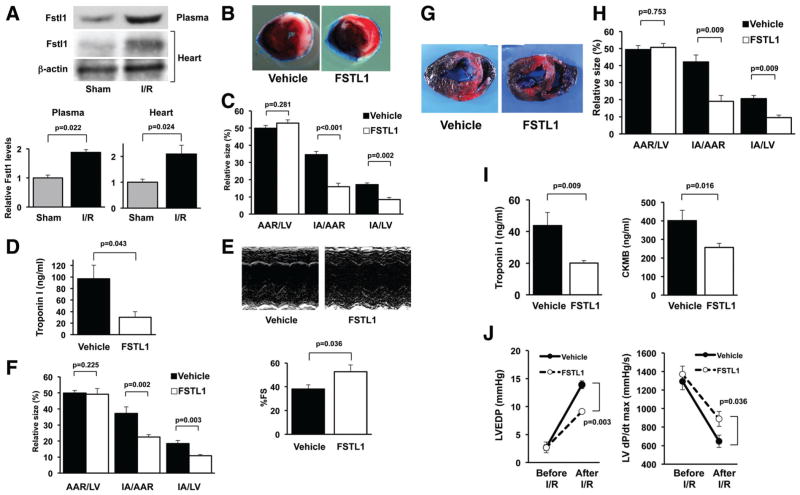
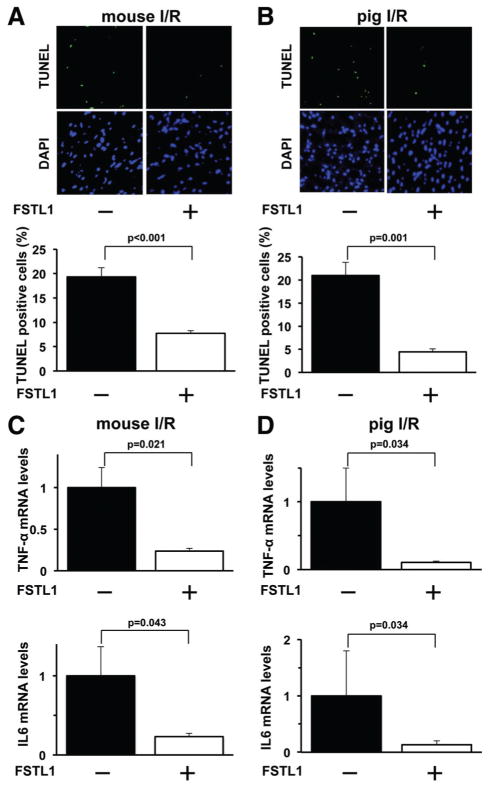
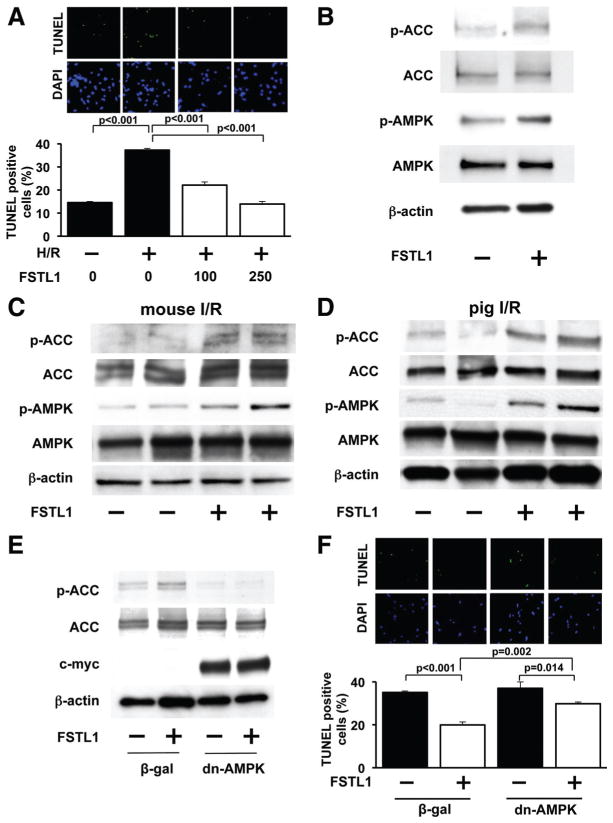
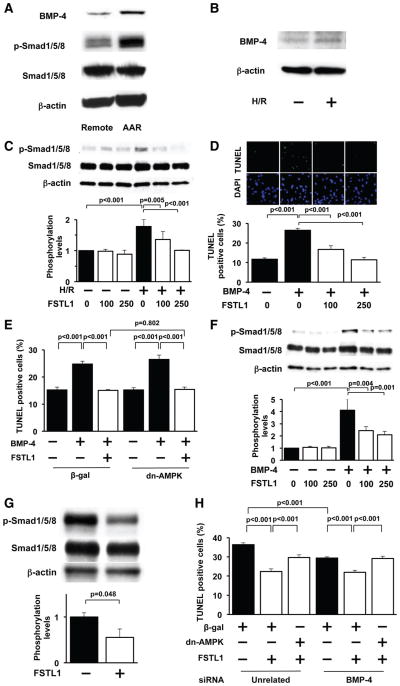
Methods and results: Administration of human FSTL1 protein significantly attenuated myocardial infarct size in a mouse or pig model of ischemia/reperfusion, which was associated with a reduction of apoptosis and inflammatory responses in the ischemic heart. Administration of FSTL1 enhanced the phosphorylation of AMP-activated protein kinase in the ischemia/reperfusion-injured heart. In cultured cardiac myocytes, FSTL1 suppressed apoptosis in response to hypoxia/reoxygenation and lipopolysaccharide-stimulated expression of proinflammatory genes through its ability to activate AMP-activated protein kinase. Ischemia/reperfusion led to enhancement of bone morphogenetic protein-4 expression and Smad1/5/8 phosphorylation in the heart, and FSTL1 suppressed the increased phosphorylation of Smad1/5/8 in ischemic myocardium. Treating cardiac myocytes with FSTL1 abolished the bone morphogenetic protein-4-stimulated increase in apoptosis, Smad1/5/8 phosphorylation, and proinflammatory gene expression. In cultured macrophages, FSTL1 diminished lipopolysaccharide-stimulated expression of proinflammatory genes via activation of AMP-activated protein kinase and abolished bone morphogenetic protein-4-dependent induction of proinflammatory mediators.
Conclusions: Our data indicate that FSTL1 can prevent myocardial ischemia/reperfusion injury by inhibiting apoptosis and inflammatory response through modulation of AMP-activated protein kinase- and bone morphogenetic protein-4-dependent mechanisms, suggesting that FSTL1 could represent a novel therapeutic target for post-myocardial infarction, acute coronary syndrome.
Figures
References
-
- Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, Carnethon MR, Dai S, de Simone G, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Greenlund KJ, Hailpern SM, Heit JA, Ho PM, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, McDermott MM, Meigs JB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Rosamond WD, Sorlie PD, Stafford RS, Turan TN, Turner MB, Wong ND, Wylie-Rosett J. Heart disease and stroke statistics—2011 update: a report from the American Heart Association. Circulation. 2011;123:e18– e209. - PMC - PubMed
-
- Lange RA, Hillis LD. Immediate angioplasty for acute myocardial infarction. N Engl J Med. 1993;328:726–728. - PubMed
-
- Fox KA, Steg PG, Eagle KA, Goodman SG, Anderson FA, Jr, Granger CB, Flather MD, Budaj A, Quill A, Gore JM. Decline in rates of death and heart failure in acute coronary syndromes, 1999 –2006. JAMA. 2007;297:1892–1900. - PubMed
-
- Simoons ML, Windecker S. Controversies in cardiovascular medicine: chronic stable coronary artery disease: drugs vs. revascularization. Eur Heart J. 2010;31:530–541. - PubMed
-
- Verma S, Fedak PW, Weisel RD, Butany J, Rao V, Maitland A, Li RK, Dhillon B, Yau TM. Fundamentals of reperfusion injury for the clinical cardiologist. Circulation. 2002;105:2332–2336. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
