

Tuning of EAG K(+) channel inactivation: molecular determinants of amplification by mutations and a small molecule
- PMID: 22930803
- PMCID: PMC3434097
- DOI: 10.1085/jgp.201210826
Tuning of EAG K(+) channel inactivation: molecular determinants of amplification by mutations and a small molecule
Abstract
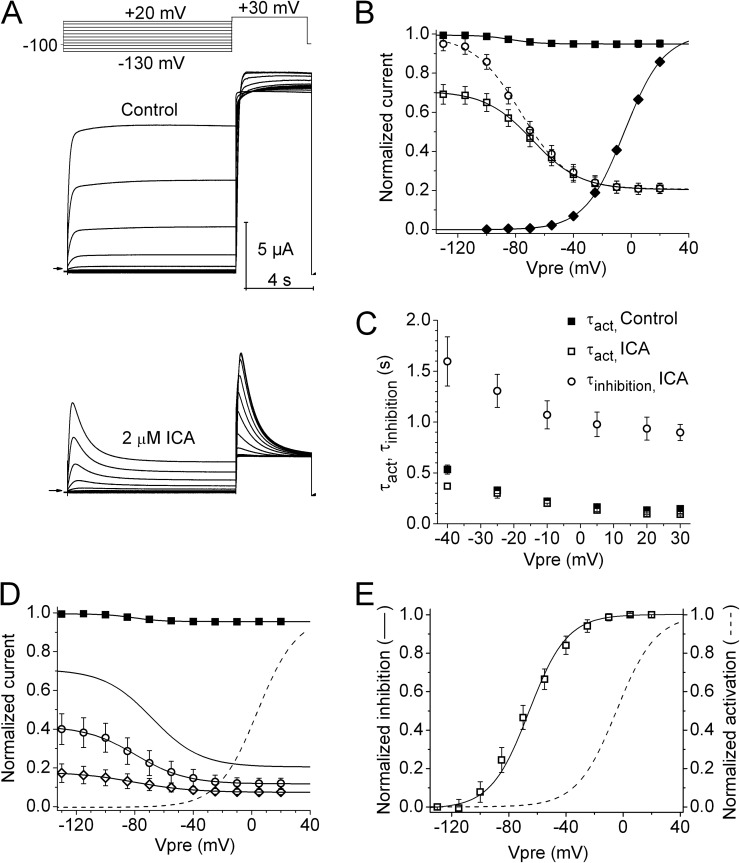
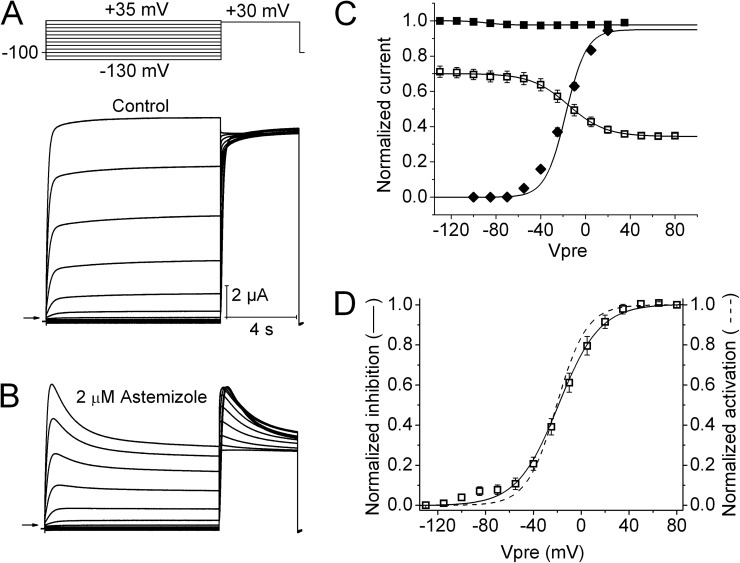
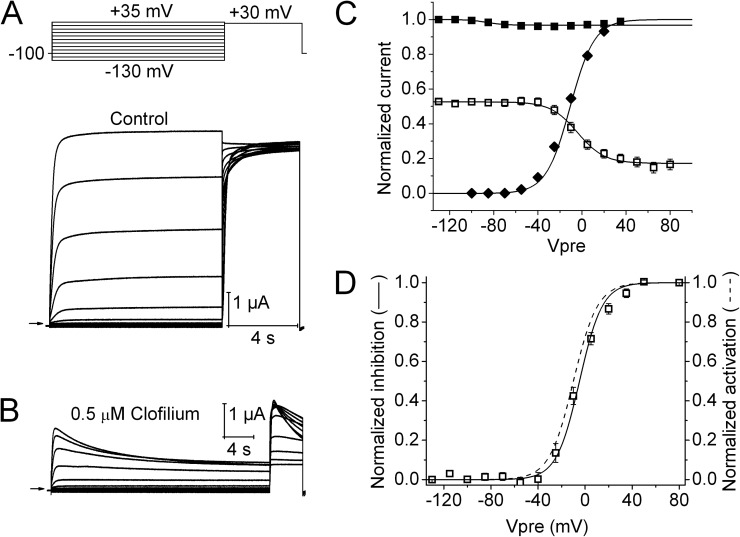
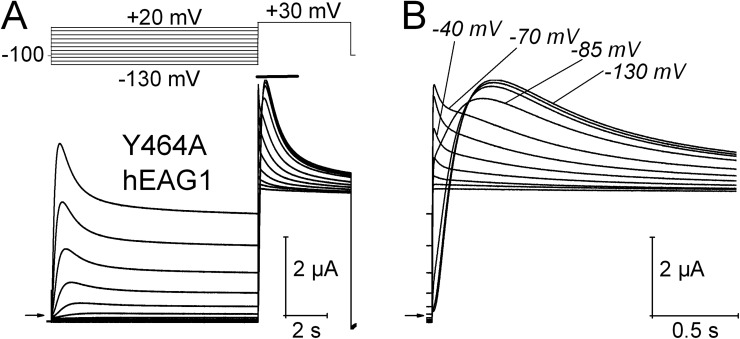
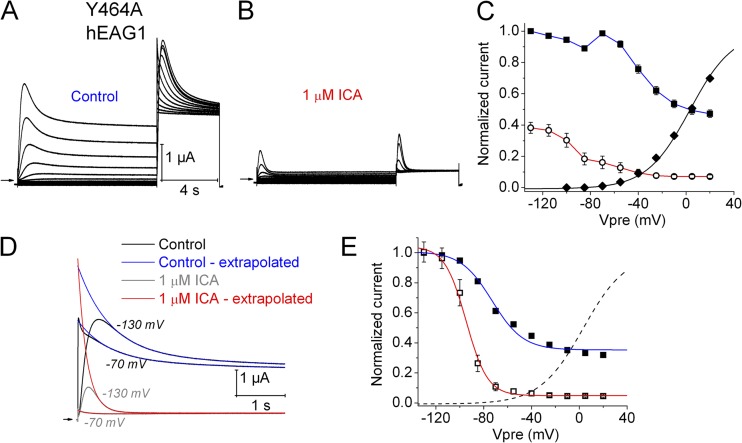
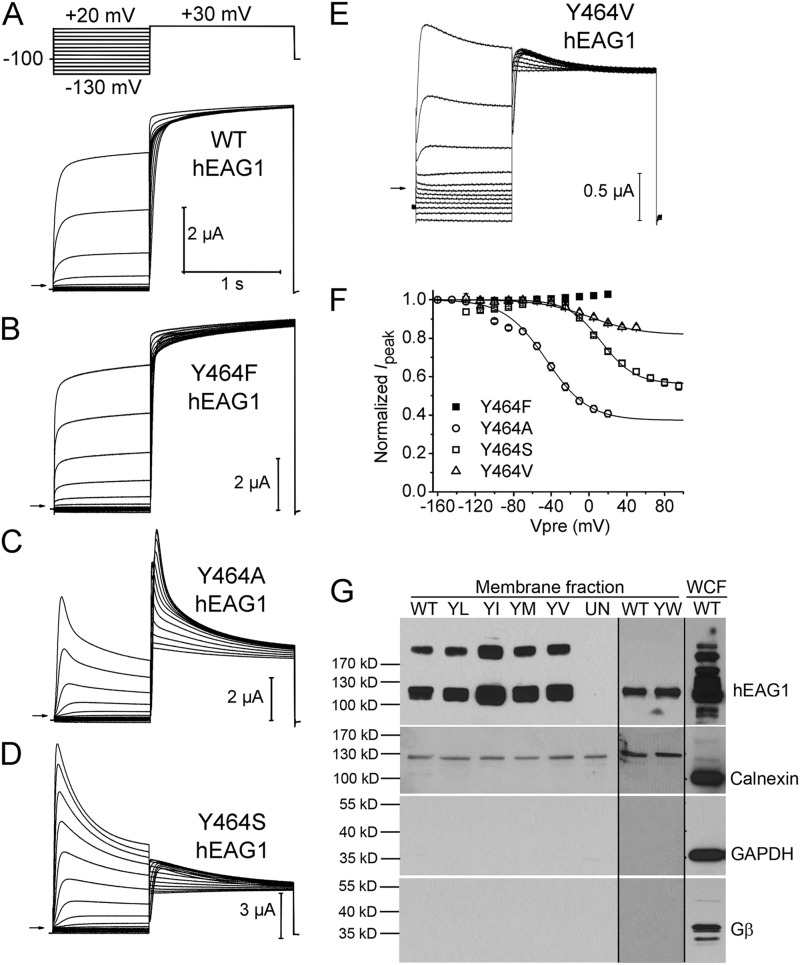
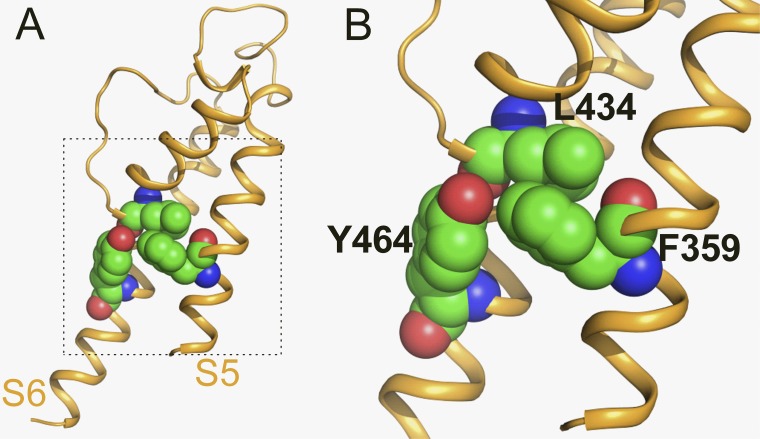
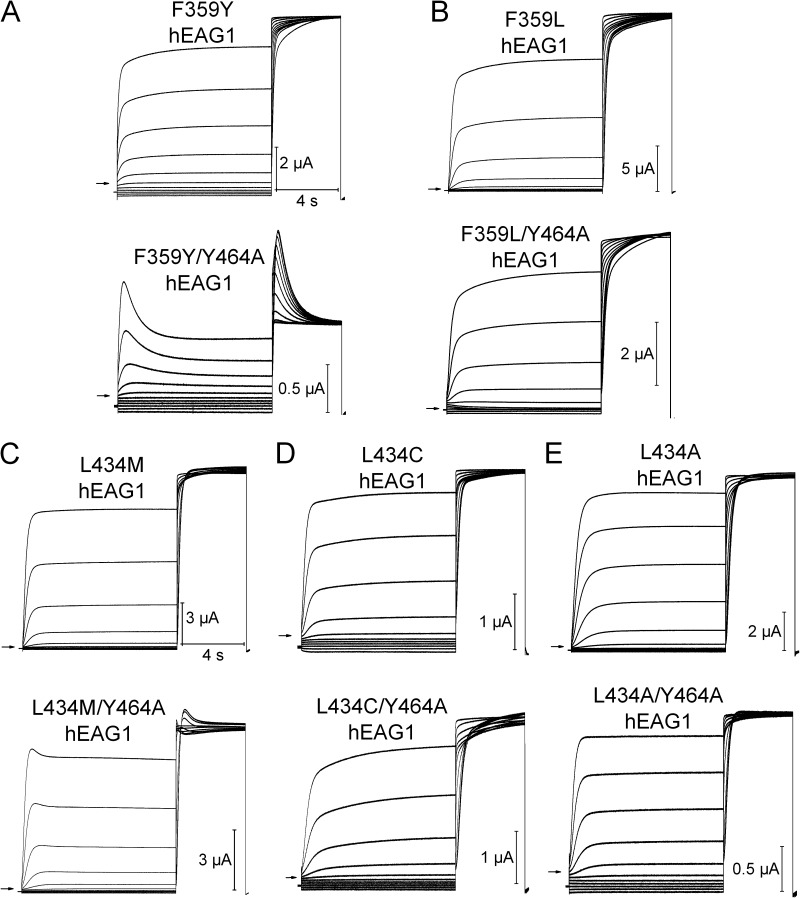
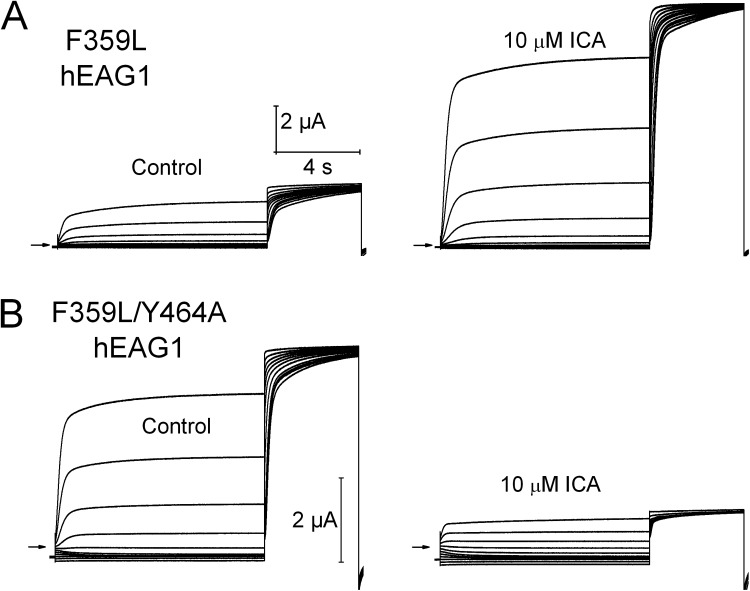
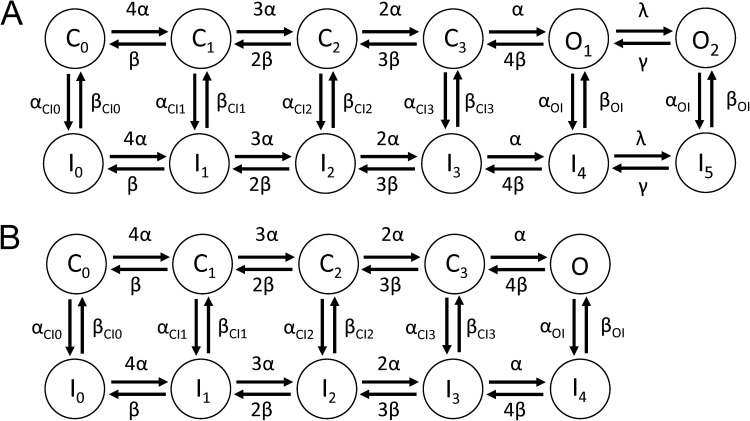
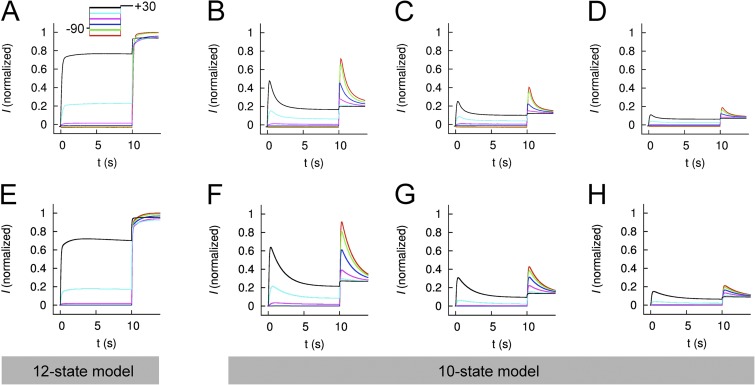
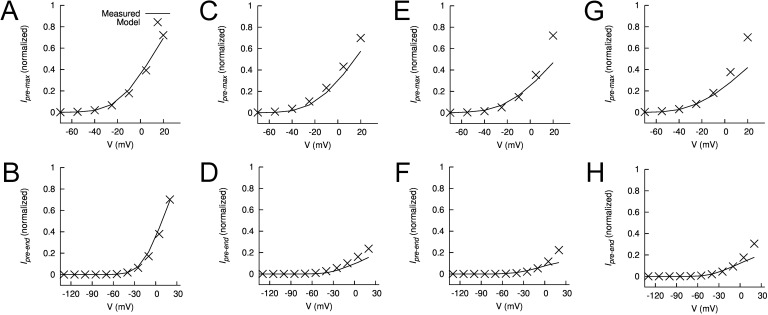
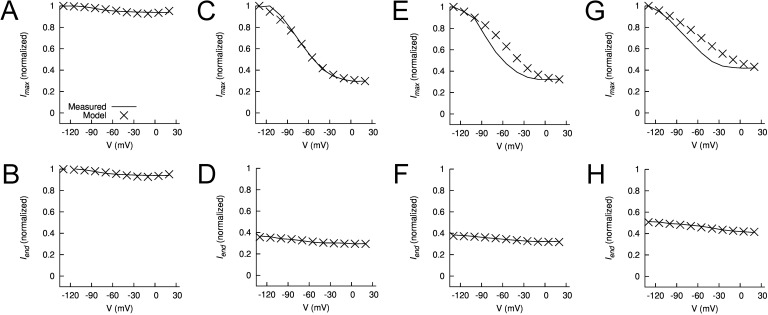
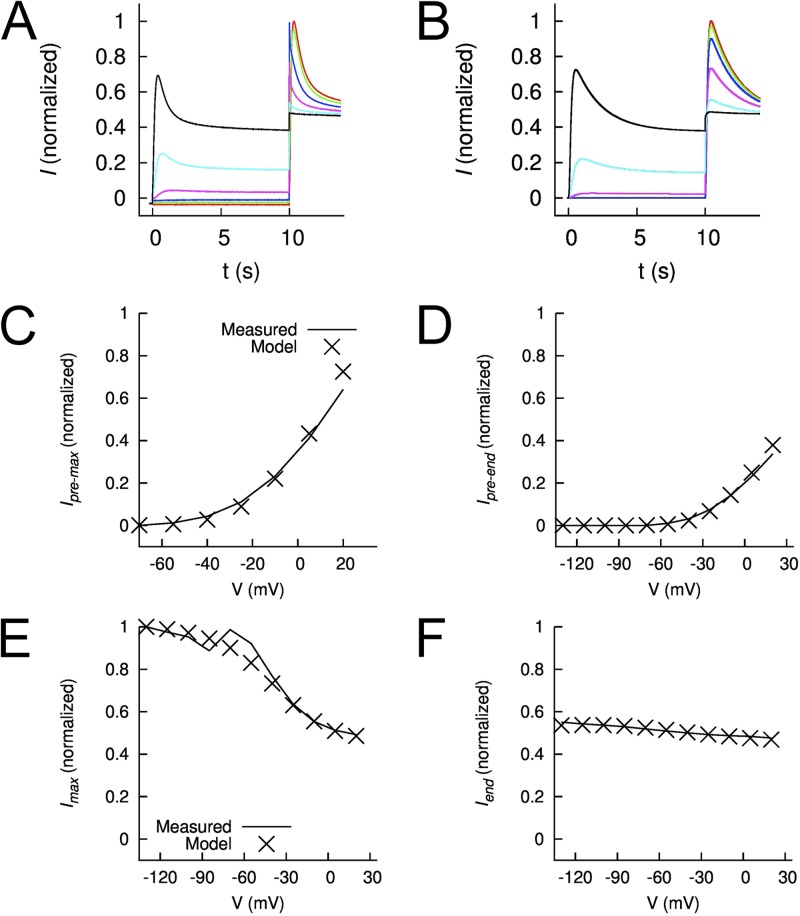
Ether-à-go-go (EAG) and EAG-related gene (ERG) K(+) channels are close homologues but differ markedly in their gating properties. ERG1 channels are characterized by rapid and extensive C-type inactivation, whereas mammalian EAG1 channels were previously considered noninactivating. Here, we show that human EAG1 channels exhibit an intrinsic voltage-dependent slow inactivation that is markedly enhanced in rate and extent by 1-10 µM 3-nitro-N-(4-phenoxyphenyl) benzamide, or ICA105574 (ICA). This compound was previously reported to have the opposite effect on ERG1 channels, causing an increase in current magnitude by inhibition of C-type inactivation. The voltage dependence of 2 µM ICA-induced inhibition of EAG1 current was half-maximal at -73 mV, 62 mV negative to the half-point for channel activation. This finding suggests that current inhibition by the drug is mediated by enhanced inactivation and not open-channel block, where the voltage half-points for current inhibition and channel activation are predicted to overlap, as we demonstrate for clofilium and astemizole. The mutation Y464A in the S6 segment also induced inactivation of EAG1, with a time course and voltage dependence similar to that caused by 2 µM ICA. Several Markov models were investigated to describe gating effects induced by multiple concentrations of the drug and the Y464A mutation. Models with the smallest fit error required both closed- and open-state inactivation. Unlike typical C-type inactivation, the rate of Y464A- and ICA-induced inactivation was not decreased by external tetraethylammonium or elevated [K(+)](e). EAG1 channel inactivation introduced by Y464A was prevented by additional mutation of a nearby residue located in the S5 segment (F359A) or pore helix (L434A), suggesting a tripartite molecular model where interactions between single residues in S5, S6, and the pore helix modulate inactivation of EAG1 channels.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
