

High variety of known and new RNA and DNA viruses of diverse origins in untreated sewage
- PMID: 22933275
- PMCID: PMC3486453
- DOI: 10.1128/JVI.00869-12
High variety of known and new RNA and DNA viruses of diverse origins in untreated sewage
Abstract
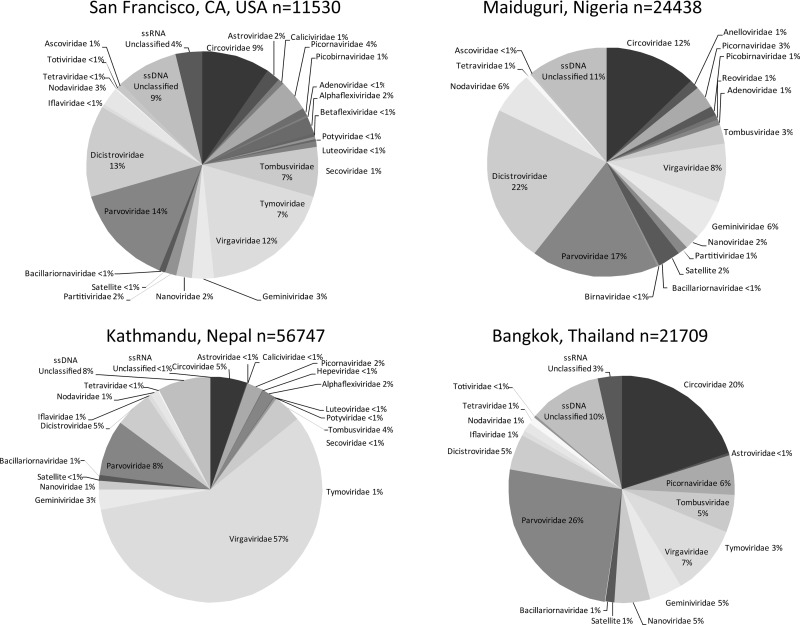
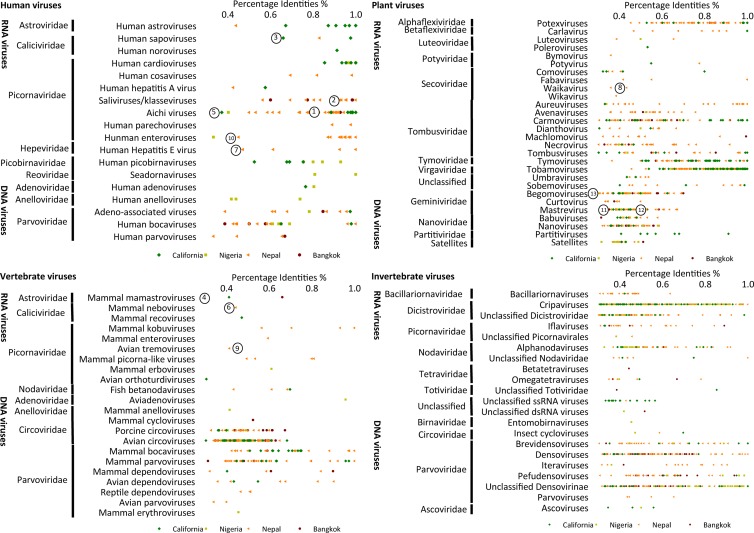
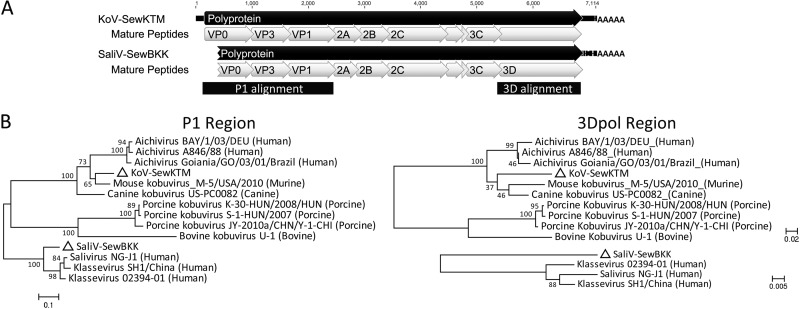
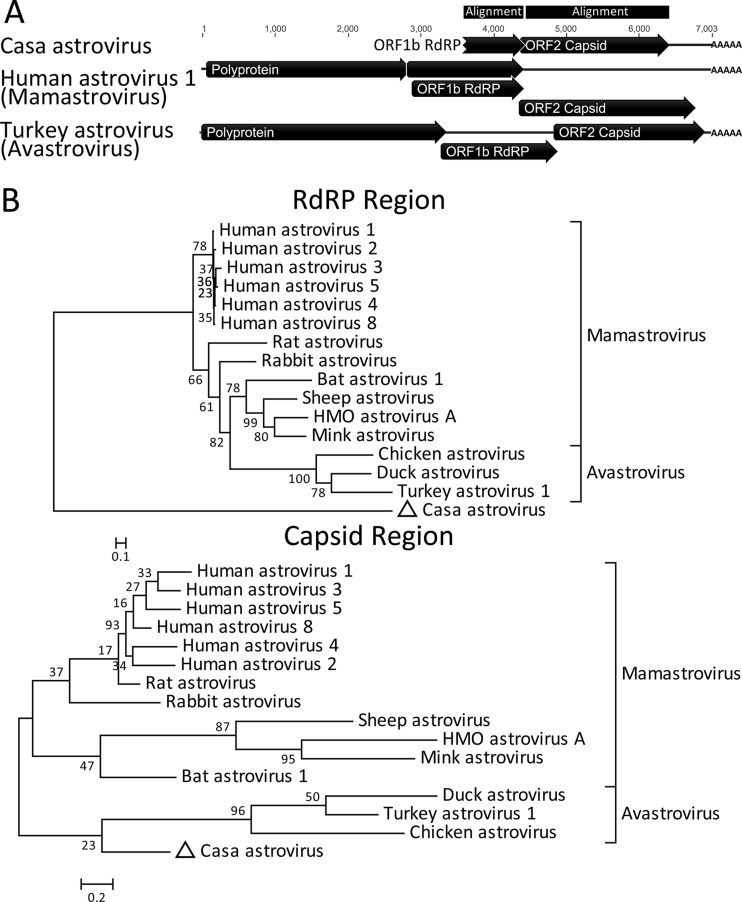
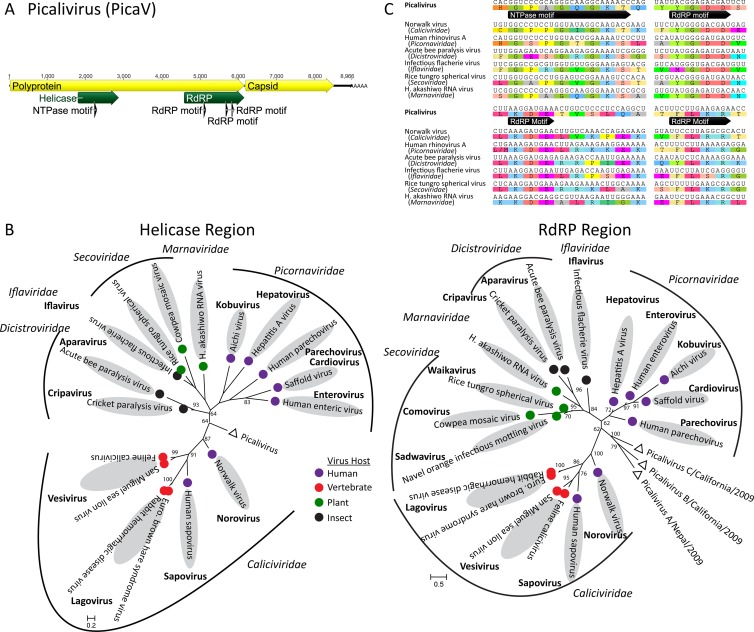
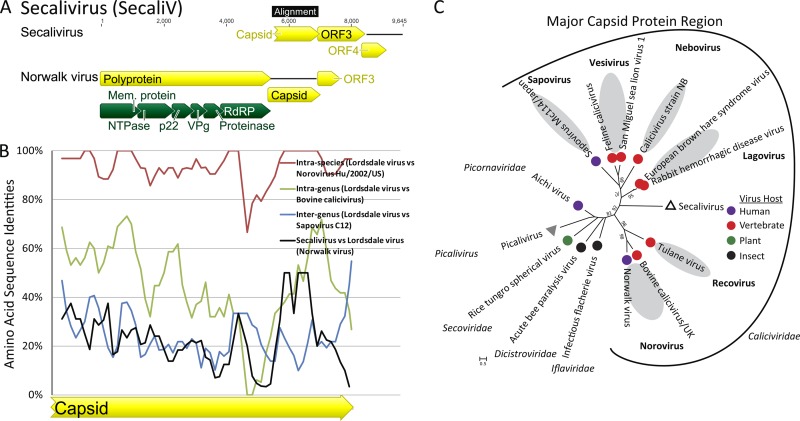
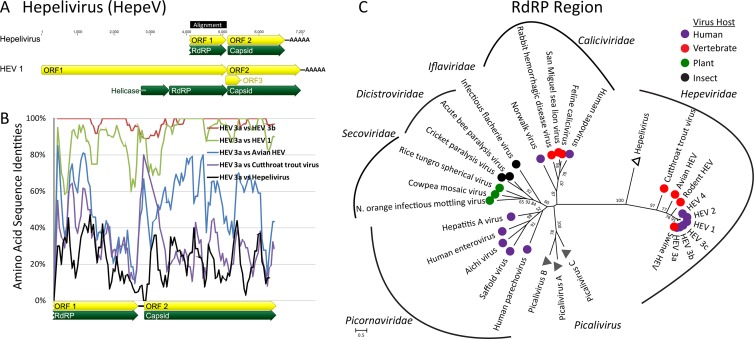
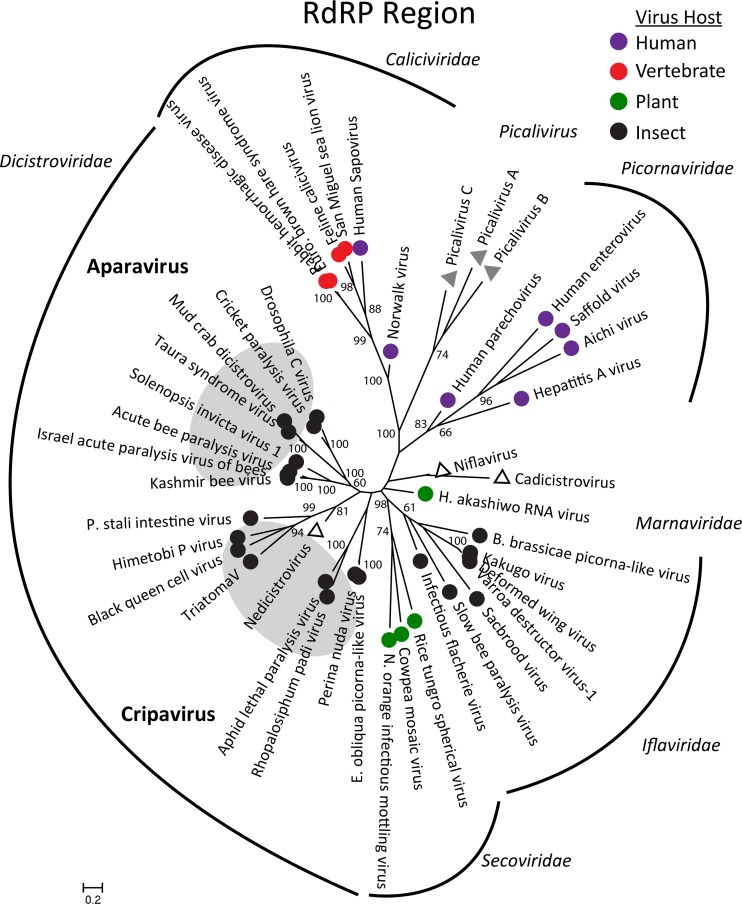
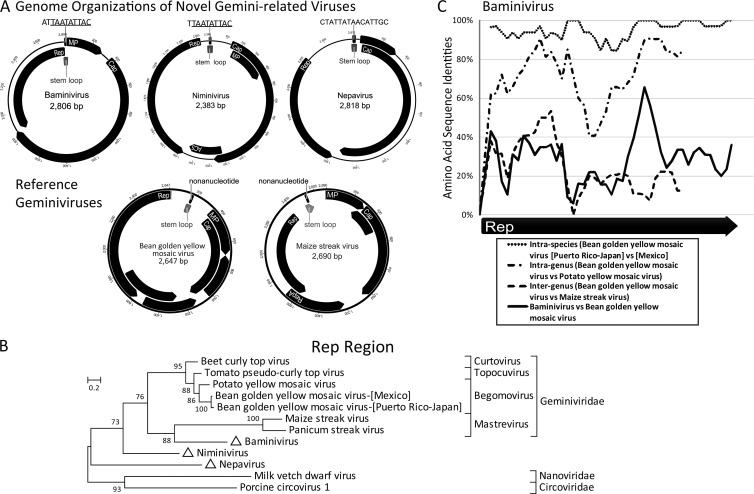
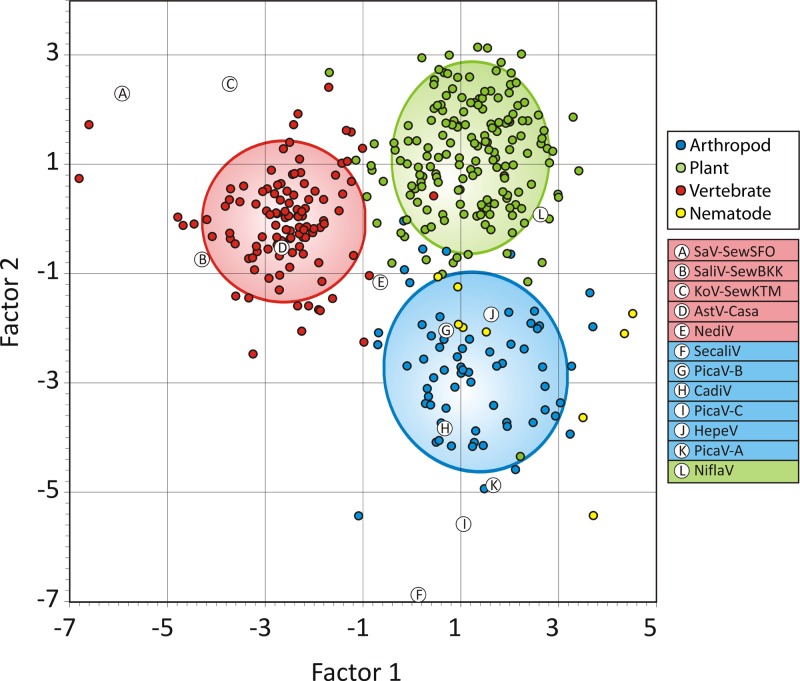
Deep sequencing of untreated sewage provides an opportunity to monitor enteric infections in large populations and for high-throughput viral discovery. A metagenomics analysis of purified viral particles in untreated sewage from the United States (San Francisco, CA), Nigeria (Maiduguri), Thailand (Bangkok), and Nepal (Kathmandu) revealed sequences related to 29 eukaryotic viral families infecting vertebrates, invertebrates, and plants (BLASTx E score, <10(-4)), including known pathogens (>90% protein identities) in numerous viral families infecting humans (Adenoviridae, Astroviridae, Caliciviridae, Hepeviridae, Parvoviridae, Picornaviridae, Picobirnaviridae, and Reoviridae), plants (Alphaflexiviridae, Betaflexiviridae, Partitiviridae, Sobemovirus, Secoviridae, Tombusviridae, Tymoviridae, Virgaviridae), and insects (Dicistroviridae, Nodaviridae, and Parvoviridae). The full and partial genomes of a novel kobuvirus, salivirus, and sapovirus are described. A novel astrovirus (casa astrovirus) basal to those infecting mammals and birds, potentially representing a third astrovirus genus, was partially characterized. Potential new genera and families of viruses distantly related to members of the single-stranded RNA picorna-like virus superfamily were genetically characterized and named Picalivirus, Secalivirus, Hepelivirus, Nedicistrovirus, Cadicistrovirus, and Niflavirus. Phylogenetic analysis placed these highly divergent genomes near the root of the picorna-like virus superfamily, with possible vertebrate, plant, or arthropod hosts inferred from nucleotide composition analysis. Circular DNA genomes distantly related to the plant-infecting Geminiviridae family were named Baminivirus, Nimivirus, and Niminivirus. These results highlight the utility of analyzing sewage to monitor shedding of viral pathogens and the high viral diversity found in this common pollutant and provide genetic information to facilitate future studies of these newly characterized viruses.
Figures
References
-
- Arthur JL, Higgins GD, Davidson GP, Givney RC, Ratcliff RM. 2009. A novel bocavirus associated with acute gastroenteritis in Australian children. PLoS Pathog. 5:e1000391 doi:10.1371/journal.ppat.1000391 - DOI - PMC - PubMed
-
- Batts W, Yun S, Hedrick R, Winton J. 2011. A novel member of the family Hepeviridae from cutthroat trout (Oncorhynchus clarkii). Virus Res. 158:116–123 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
