

The role of bacterial enhancer binding proteins as specialized activators of σ54-dependent transcription
- PMID: 22933558
- PMCID: PMC3429621
- DOI: 10.1128/MMBR.00006-12
The role of bacterial enhancer binding proteins as specialized activators of σ54-dependent transcription
Abstract
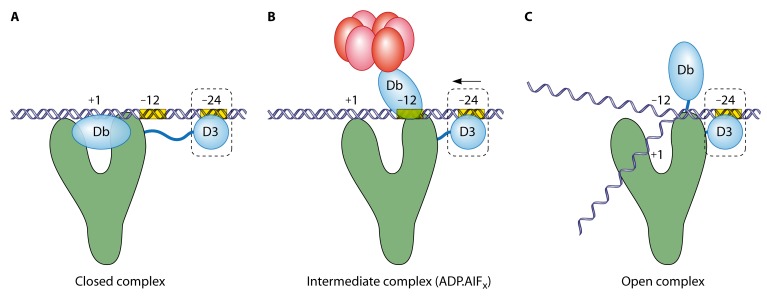
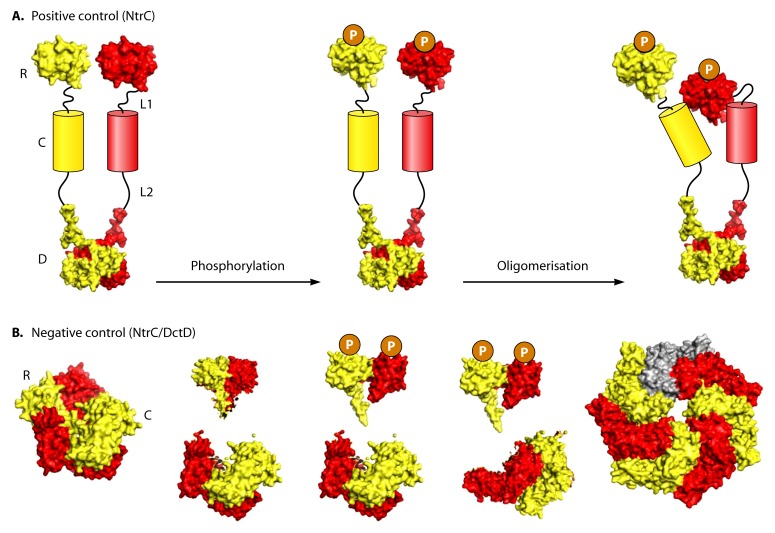
Bacterial enhancer binding proteins (bEBPs) are transcriptional activators that assemble as hexameric rings in their active forms and utilize ATP hydrolysis to remodel the conformation of RNA polymerase containing the alternative sigma factor σ(54). We present a comprehensive and detailed summary of recent advances in our understanding of how these specialized molecular machines function. The review is structured by introducing each of the three domains in turn: the central catalytic domain, the N-terminal regulatory domain, and the C-terminal DNA binding domain. The role of the central catalytic domain is presented with particular reference to (i) oligomerization, (ii) ATP hydrolysis, and (iii) the key GAFTGA motif that contacts σ(54) for remodeling. Each of these functions forms a potential target of the signal-sensing N-terminal regulatory domain, which can act either positively or negatively to control the activation of σ(54)-dependent transcription. Finally, we focus on the DNA binding function of the C-terminal domain and the enhancer sites to which it binds. Particular attention is paid to the importance of σ(54) to the bacterial cell and its unique role in regulating transcription.
Figures


















References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
