

Genetic dysfunction of MT-ATP6 causes axonal Charcot-Marie-Tooth disease
- PMID: 22933740
- PMCID: PMC3525307
- DOI: 10.1212/WNL.0b013e3182698d8d
Genetic dysfunction of MT-ATP6 causes axonal Charcot-Marie-Tooth disease
Abstract
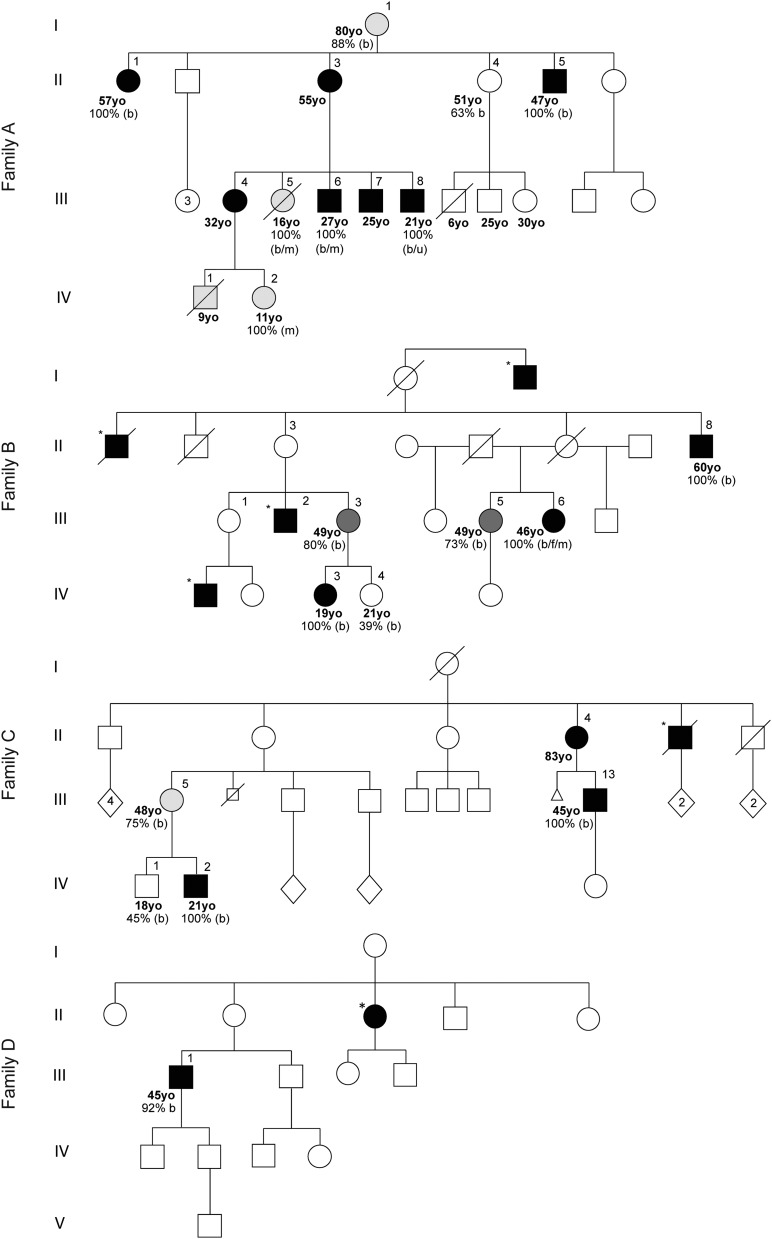
Objective: Charcot-Marie-Tooth (CMT) disease is the most common inherited neuromuscular disorder, affecting 1 in 2,500 individuals. Mitochondrial DNA (mtDNA) mutations are not generally considered within the differential diagnosis of patients with uncomplicated inherited neuropathy, despite the essential requirement of ATP for axonal function. We identified the mtDNA mutation m.9185T>C in MT-ATP6, encoding the ATP6 subunit of the mitochondrial ATP synthase (OXPHOS complex V), at homoplasmic levels in a family with mitochondrial disease in whom a severe motor axonal neuropathy was a striking feature. This led us to hypothesize that mutations in the 2 mtDNA complex V subunit encoding genes, MT-ATP6 and MT-ATP8, might be an unrecognized cause of isolated axonal CMT and distal hereditary motor neuropathy (dHMN).
Methods: A total of 442 probands with CMT type 2 (CMT2) (270) and dHMN (172) were screened for MT-ATP6/8 mutations after exclusion of mutations in known CMT2/dHMN genes. Mutation load was quantified using restriction endonuclease analysis. Blue-native gel electrophoresis (BN-PAGE) was performed to analyze the effects of m.9185T>C on complex V structure and function.
Results: Three further probands with CMT2 harbored the m.9185T>C mutation. Some relatives had been classified as having dHMN. Patients could be separated into 4 groups according to their mutant m.9185T>C levels. BN-PAGE demonstrated both impaired assembly and reduced activity of the complex V holoenzyme.
Conclusions: We have shown that m.9185T>C in MT-ATP6 causes CMT2 in 1.1% of genetically undefined cases. This has important implications for diagnosis and genetic counseling. Recognition that mutations in MT-ATP6 cause CMT2 enhances current understanding of the pathogenic basis of axonal neuropathy.
Figures

References
-
- Pezeshkpour G, Krarup C, Buchthal F, et al. Peripheral neuropathy in mitochondrial disease. J Neurol Sci 1987;77:285–304 - PubMed
-
- Coker SB. Leigh disease presenting as Guillain-Barré syndrome. Pediatr Neurol 1993;9:61–63 - PubMed
-
- Rahman S, Blok RB, Dahl HH, et al. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol 1996;39:343–351 - PubMed
-
- Chu CC, Huang CC, Fang W, et al. Peripheral neuropathy in mitochondrial encephalomyopathies. Eur Neurol 1997;37:110–115 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical