

Adaptor protein APPL1 couples synaptic NMDA receptor with neuronal prosurvival phosphatidylinositol 3-kinase/Akt pathway
- PMID: 22933778
- PMCID: PMC6621526
- DOI: 10.1523/JNEUROSCI.3852-11.2012
Adaptor protein APPL1 couples synaptic NMDA receptor with neuronal prosurvival phosphatidylinositol 3-kinase/Akt pathway
Abstract
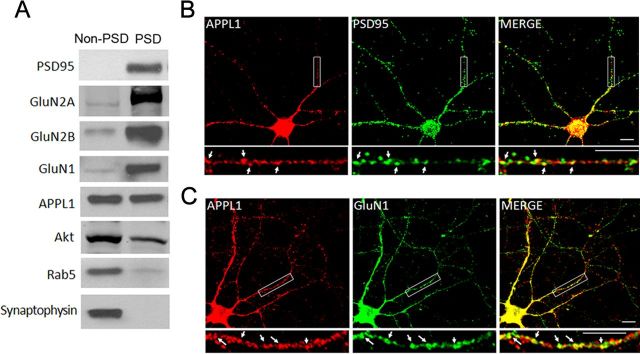
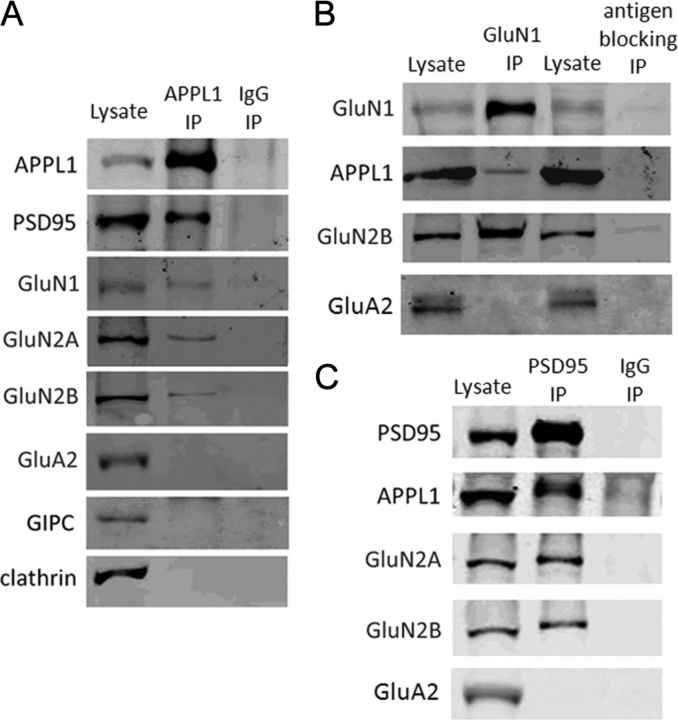
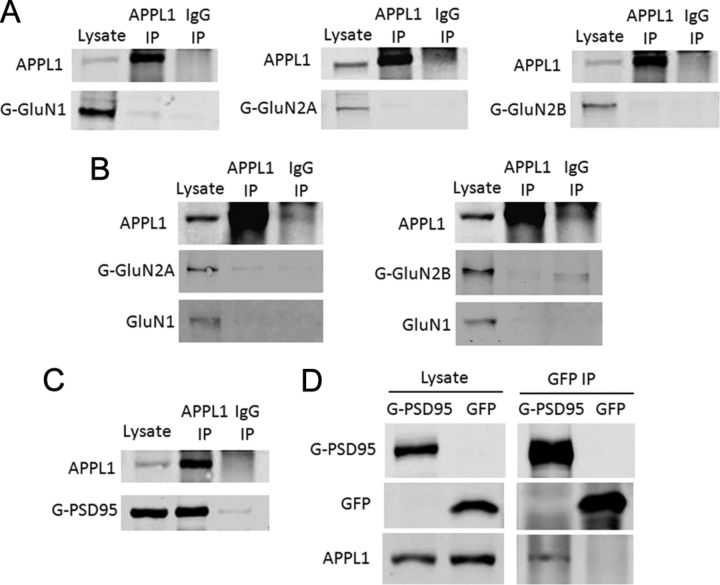
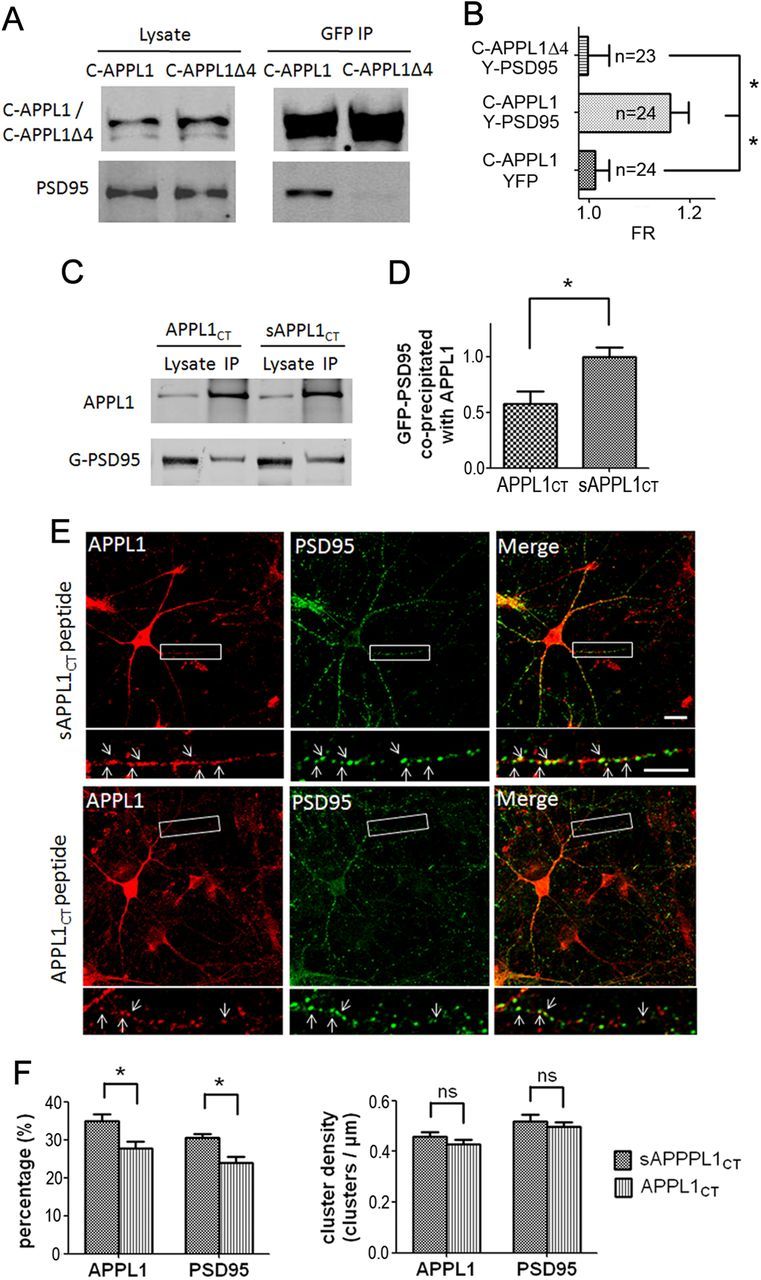
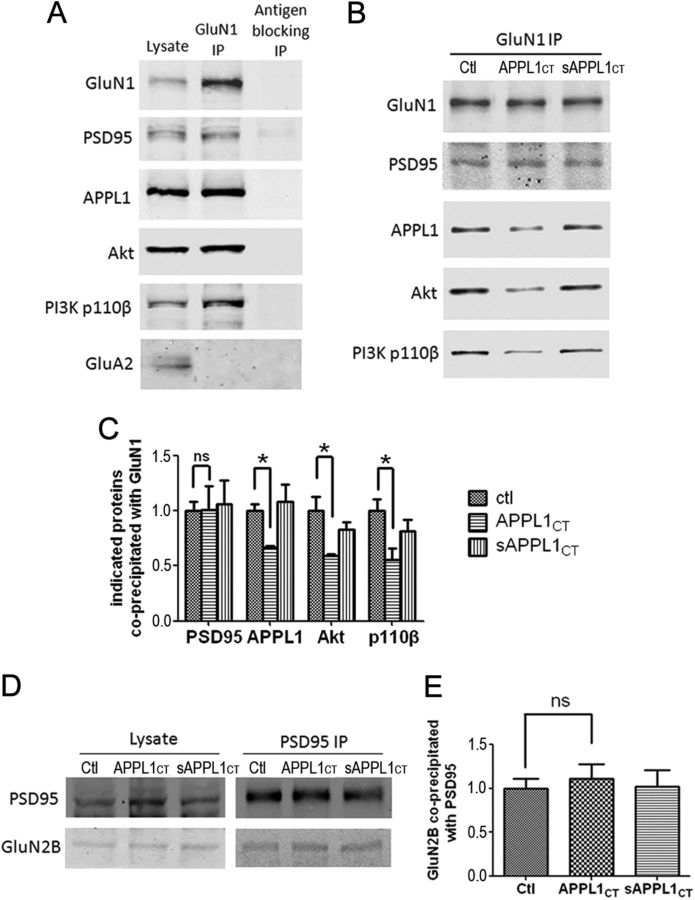
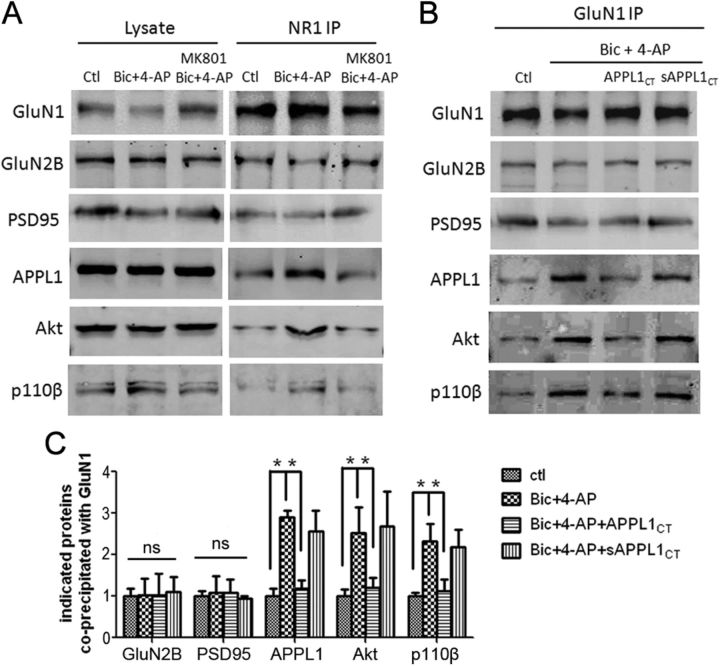
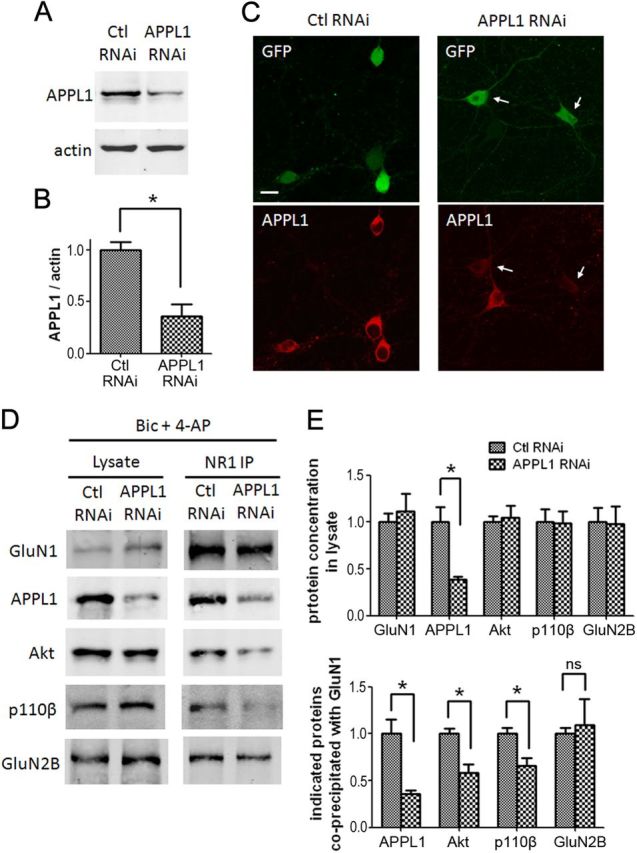
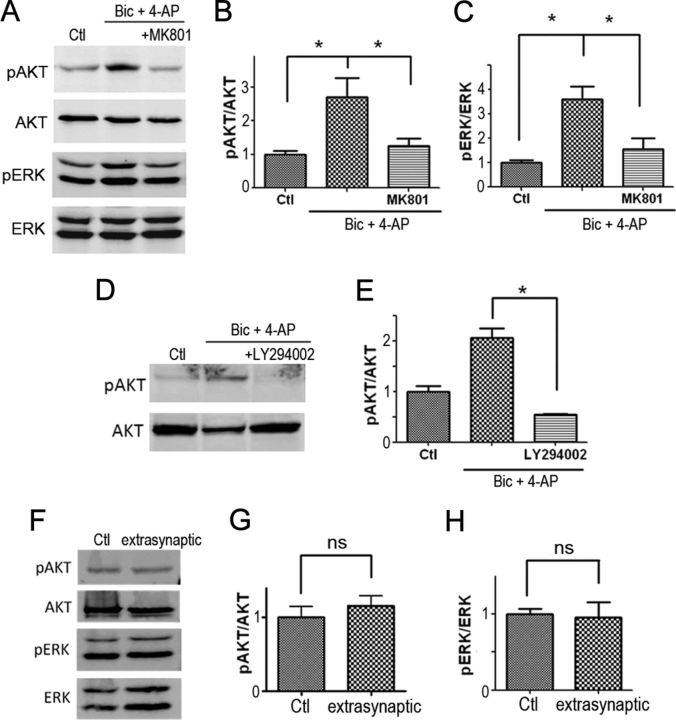
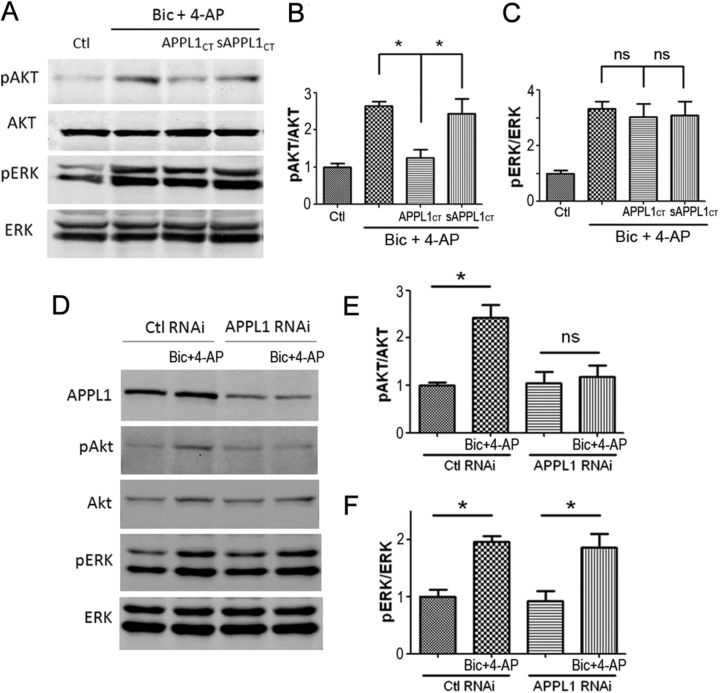
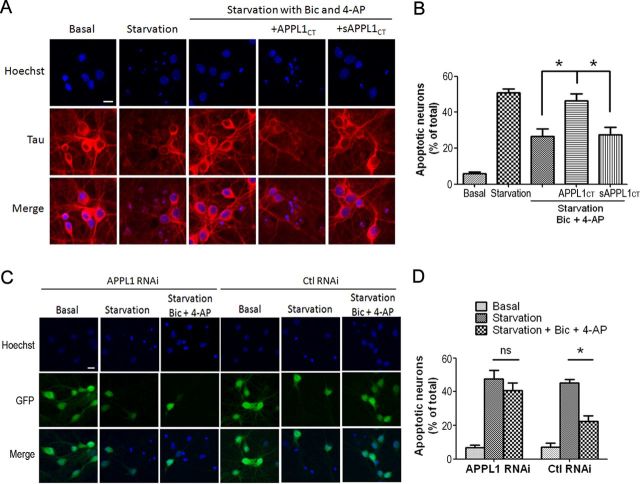
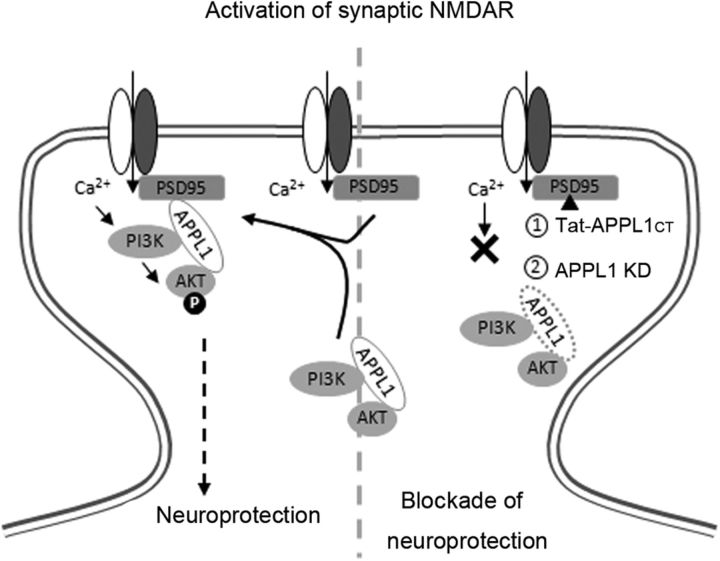
It is well known that NMDA receptors (NMDARs) can both induce neurotoxicity and promote neuronal survival under different circumstances. Recent studies show that such paradoxical responses are related to the receptor location: the former to the extrasynaptic and the latter to the synaptic. The phosphoinositide 3-kinase (PI3K)/Akt kinase cascade is a key pathway responsible for the synaptic NMDAR-dependent neuroprotection. However, it is still unknown how synaptic NMDARs are coupled with the PI3K/Akt pathway. Here, we explored the role of an adaptor protein-adaptor protein containing pH domain, PTB domain, and leucine zipper motif (APPL1)-in this signal coupling using rat cortical neurons. We found that APPL1 existed in postsynaptic densities and associated with the NMDAR complex through binding to PSD95 at its C-terminal PDZ-binding motif. NMDARs, APPL1, and the PI3K/Akt cascade formed a complex in rat cortical neurons. Synaptic NMDAR activity increased the association of this complex, induced activation of the PI3K/Akt pathway, and consequently protected neurons against starvation-induced apoptosis. Perturbing APPL1 interaction with PSD95 by a peptide comprising the APPL1 C-terminal PDZ-binding motif dissociated the PI3K/Akt pathway from NMDARs. Either the peptide or lentiviral knockdown of APPL1 blocked synaptic NMDAR-dependent recruitment and activation of PI3K/Akt pathway, and consequently blocked synaptic NMDAR-dependent neuroprotection. These results suggest that APPL1 contributes to connecting synaptic NMDARs with the intracellular PI3K/Akt cascade and the downstream prosurvival signaling pathway in rat cortical neurons.
Figures
Similar articles
-
Serine 707 of APPL1 is Critical for the Synaptic NMDA Receptor-Mediated Akt Phosphorylation Signaling Pathway.Neurosci Bull. 2016 Aug;32(4):323-30. doi: 10.1007/s12264-016-0042-9. Epub 2016 Jun 14. Neurosci Bull. 2016. PMID: 27300007 Free PMC article.
-
An APPL1/Akt signaling complex regulates dendritic spine and synapse formation in hippocampal neurons.Mol Cell Neurosci. 2011 Mar;46(3):633-44. doi: 10.1016/j.mcn.2011.01.003. Epub 2011 Jan 12. Mol Cell Neurosci. 2011. PMID: 21236345 Free PMC article.
-
APPL1 gates long-term potentiation through its plekstrin homology domain.J Cell Sci. 2016 Jul 15;129(14):2793-803. doi: 10.1242/jcs.183475. Epub 2016 Jun 2. J Cell Sci. 2016. PMID: 27257087
-
Regulation of neuronal PKA signaling through AKAP targeting dynamics.Eur J Cell Biol. 2006 Jul;85(7):627-33. doi: 10.1016/j.ejcb.2006.01.010. Epub 2006 Feb 28. Eur J Cell Biol. 2006. PMID: 16504338 Review.
-
NMDA receptor activity determines neuronal fate: location or number?Rev Neurosci. 2015;26(1):39-47. doi: 10.1515/revneuro-2014-0053. Rev Neurosci. 2015. PMID: 25324444 Review.
Cited by
-
Serine 707 of APPL1 is Critical for the Synaptic NMDA Receptor-Mediated Akt Phosphorylation Signaling Pathway.Neurosci Bull. 2016 Aug;32(4):323-30. doi: 10.1007/s12264-016-0042-9. Epub 2016 Jun 14. Neurosci Bull. 2016. PMID: 27300007 Free PMC article.
-
De novo mutations in GRIN1 cause extensive bilateral polymicrogyria.Brain. 2018 Mar 1;141(3):698-712. doi: 10.1093/brain/awx358. Brain. 2018. PMID: 29365063 Free PMC article.
-
Pregnenolone sulfate normalizes schizophrenia-like behaviors in dopamine transporter knockout mice through the AKT/GSK3β pathway.Transl Psychiatry. 2015 Mar 17;5(3):e528. doi: 10.1038/tp.2015.21. Transl Psychiatry. 2015. PMID: 25781227 Free PMC article.
-
NADPH oxidase-2: linking glucose, acidosis, and excitotoxicity in stroke.Antioxid Redox Signal. 2015 Jan 10;22(2):161-74. doi: 10.1089/ars.2013.5767. Antioxid Redox Signal. 2015. PMID: 24628477 Free PMC article.
-
Intranasal administration of recombinant Netrin-1 attenuates neuronal apoptosis by activating DCC/APPL-1/AKT signaling pathway after subarachnoid hemorrhage in rats.Neuropharmacology. 2017 Jun;119:123-133. doi: 10.1016/j.neuropharm.2017.03.025. Epub 2017 Mar 24. Neuropharmacology. 2017. PMID: 28347836 Free PMC article.
References
-
- Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, MacDonald JF, Tymianski M. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003;115:863–877. - PubMed
-
- Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261–269. - PubMed
-
- Downward J. How BAD phosphorylation is good for survival. Nat Cell Biol. 1999;1:E33–E35. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases