

The C-type natriuretic peptide induces thermal hyperalgesia through a noncanonical Gβγ-dependent modulation of TRPV1 channel
- PMID: 22933780
- PMCID: PMC3461320
- DOI: 10.1523/JNEUROSCI.1330-12.2012
The C-type natriuretic peptide induces thermal hyperalgesia through a noncanonical Gβγ-dependent modulation of TRPV1 channel
Abstract
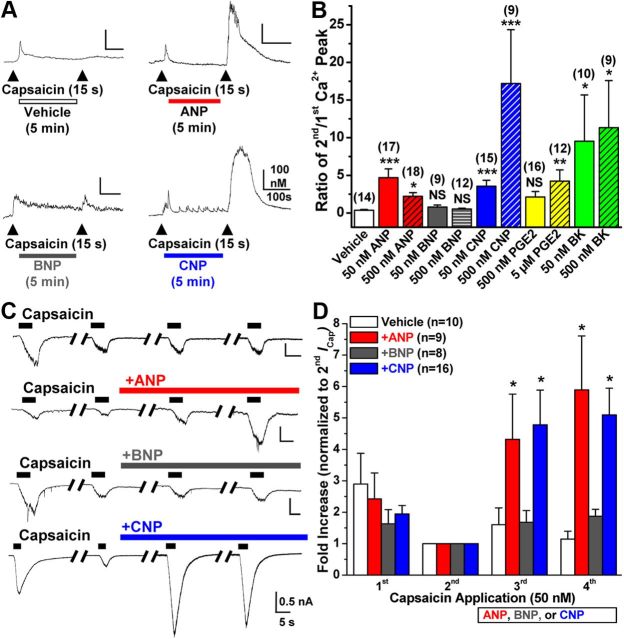
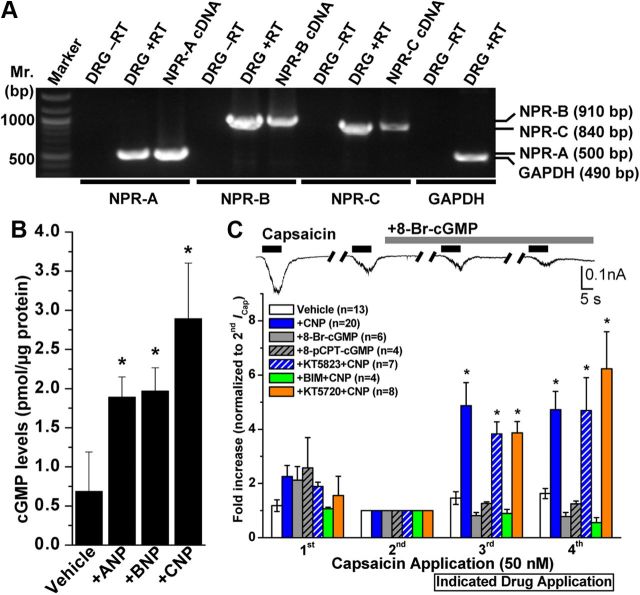
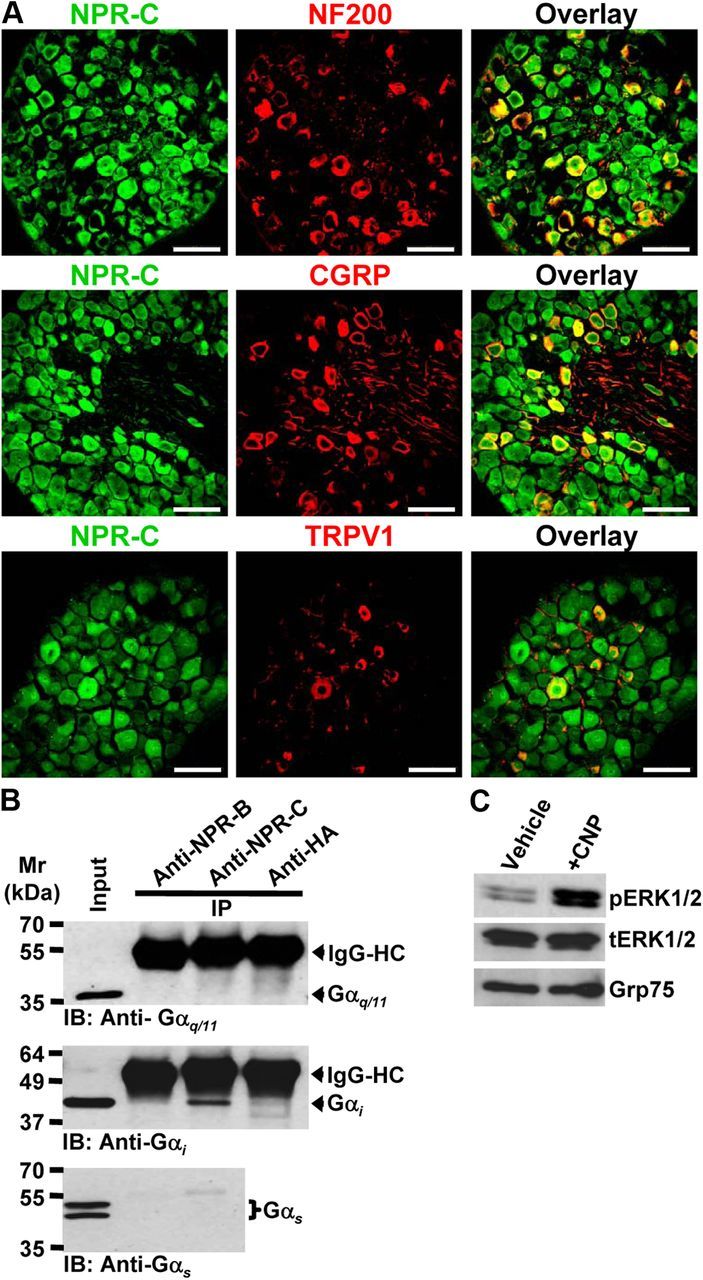
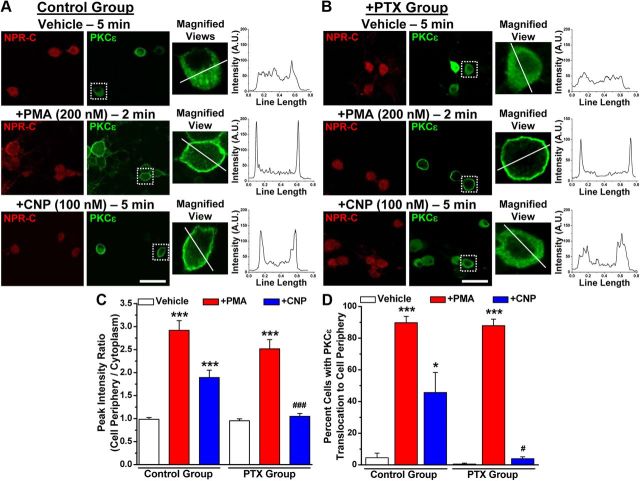
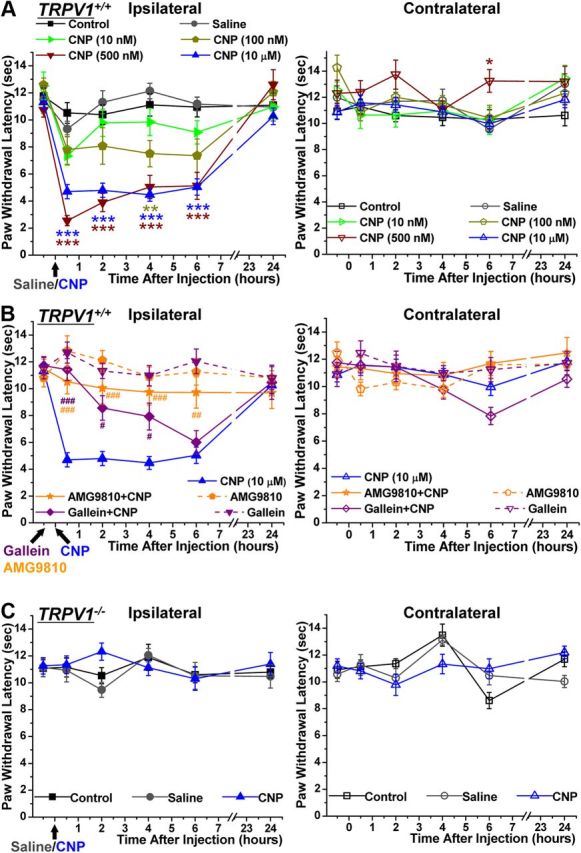
Natriuretic peptides (NPs) control natriuresis and normalize changes in blood pressure. Recent studies suggest that NPs are also involved in the regulation of pain sensitivity, although the underlying mechanisms remain essentially unknown. Many biological effects of NPs are mediated by guanylate cyclase (GC)-coupled NP receptors, NPR-A and NPR-B, whereas the third NP receptor, NPR-C, lacks the GC kinase domain and acts as the NP clearance receptor. In addition, NPR-C can couple to specific Gα(i)-Gβγ-mediated intracellular signaling cascades in numerous cell types. We found that NPR-C is coexpressed in transient receptor potential vanilloid-1 (TRPV1)-expressing mouse dorsal root ganglia (DRG) neurons. NPR-C can be coimmunoprecipitated with Gα(i), and C-type natriuretic peptide (CNP) treatment induced translocation of protein kinase Cε (PKCε) to the plasma membrane of these neurons, which was inhibited by pertussis toxin pretreatment. Application of CNP potentiated capsaicin- and proton-activated TRPV1 currents in cultured mouse DRG neurons and increased their firing frequency, an effect that was absent in DRG neurons from TRPV1(-/-) mice. CNP-induced sensitization of TRPV1 activity was attenuated by pretreatment of DRG neurons with the specific inhibitors of Gβγ, phospholipase C-β (PLCβ), or PKC, but not of protein kinase A, and was abolished by mutations at two PKC phosphorylation sites in TRPV1. Furthermore, CNP injection into mouse hindpaw led to the development of thermal hyperalgesia that was attenuated by administration of specific inhibitors of Gβγ or TRPV1 and was also absent in TRPV1(-/-) mice. Thus, our work identifies the Gβγ-PLCβ-PKC-dependent potentiation of TRPV1 as a novel signaling cascade recruited by CNP in mouse DRG neurons that can lead to enhanced nociceptor excitability and thermal hypersensitivity.
Figures
Similar articles
-
Promiscuous G-Protein-Coupled Receptor Inhibition of Transient Receptor Potential Melastatin 3 Ion Channels by Gβγ Subunits.J Neurosci. 2019 Oct 2;39(40):7840-7852. doi: 10.1523/JNEUROSCI.0882-19.2019. Epub 2019 Aug 26. J Neurosci. 2019. PMID: 31451581 Free PMC article.
-
Protease-activated receptor 2 sensitizes the capsaicin receptor transient receptor potential vanilloid receptor 1 to induce hyperalgesia.J Neurosci. 2004 May 5;24(18):4300-12. doi: 10.1523/JNEUROSCI.5679-03.2004. J Neurosci. 2004. PMID: 15128844 Free PMC article.
-
Neurokinin-1 receptor enhances TRPV1 activity in primary sensory neurons via PKCepsilon: a novel pathway for heat hyperalgesia.J Neurosci. 2007 Oct 31;27(44):12067-77. doi: 10.1523/JNEUROSCI.0496-07.2007. J Neurosci. 2007. PMID: 17978048 Free PMC article.
-
Protease-activated receptor 2 sensitizes TRPV1 by protein kinase Cepsilon- and A-dependent mechanisms in rats and mice.J Physiol. 2006 Sep 1;575(Pt 2):555-71. doi: 10.1113/jphysiol.2006.111534. Epub 2006 Jun 22. J Physiol. 2006. PMID: 16793902 Free PMC article.
-
Neurotrophic Factors and Nociceptor Sensitization.In: Kruger L, Light AR, editors. Translational Pain Research: From Mouse to Man. Boca Raton (FL): CRC Press/Taylor & Francis; 2010. Chapter 2. In: Kruger L, Light AR, editors. Translational Pain Research: From Mouse to Man. Boca Raton (FL): CRC Press/Taylor & Francis; 2010. Chapter 2. PMID: 21882462 Free Books & Documents. Review.
Cited by
-
Abnormal trigeminal sensory processing in obese mice.Pain. 2016 Jan;157(1):235-246. doi: 10.1097/j.pain.0000000000000355. Pain. 2016. PMID: 26397933 Free PMC article.
-
Itch-associated peptides: RNA-Seq and bioinformatic analysis of natriuretic precursor peptide B and gastrin releasing peptide in dorsal root and trigeminal ganglia, and the spinal cord.Mol Pain. 2014 Aug 14;10:44. doi: 10.1186/1744-8069-10-44. Mol Pain. 2014. PMID: 25123163 Free PMC article.
-
Sensory TRP channels: the key transducers of nociception and pain.Prog Mol Biol Transl Sci. 2015;131:73-118. doi: 10.1016/bs.pmbts.2015.01.002. Epub 2015 Feb 12. Prog Mol Biol Transl Sci. 2015. PMID: 25744671 Free PMC article. Review.
-
Phylogenetic Analysis Provides Insight Into the Molecular Evolution of Nociception and Pain-Related Proteins.Evol Bioinform Online. 2023 Dec 14;19:11769343231216914. doi: 10.1177/11769343231216914. eCollection 2023. Evol Bioinform Online. 2023. PMID: 38107163 Free PMC article.
-
Chemokine co-receptor CCR5/CXCR4-dependent modulation of Kv2.1 channel confers acute neuroprotection to HIV-1 glycoprotein gp120 exposure.PLoS One. 2013 Sep 24;8(9):e76698. doi: 10.1371/journal.pone.0076698. eCollection 2013. PLoS One. 2013. PMID: 24086760 Free PMC article.
References
-
- Ahluwalia A, Hobbs AJ. Endothelium-derived C-type natriuretic peptide: more than just a hyperpolarizing factor. Trends Pharmacol Sci. 2005;26:162–167. - PubMed
-
- Anand-Srivastava MB. Natriuretic peptide receptor-C signaling and regulation. Peptides. 2005;26:1044–1059. - PubMed
-
- Bhave G, Gereau RW., 4th Posttranslational mechanisms of peripheral sensitization. J Neurobiol. 2004;61:88–106. - PubMed
-
- Bhave G, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RW., 4th cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron. 2002;35:721–731. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous