

Integration of expression data in genome-scale metabolic network reconstructions
- PMID: 22934050
- PMCID: PMC3429070
- DOI: 10.3389/fphys.2012.00299
Integration of expression data in genome-scale metabolic network reconstructions
Abstract
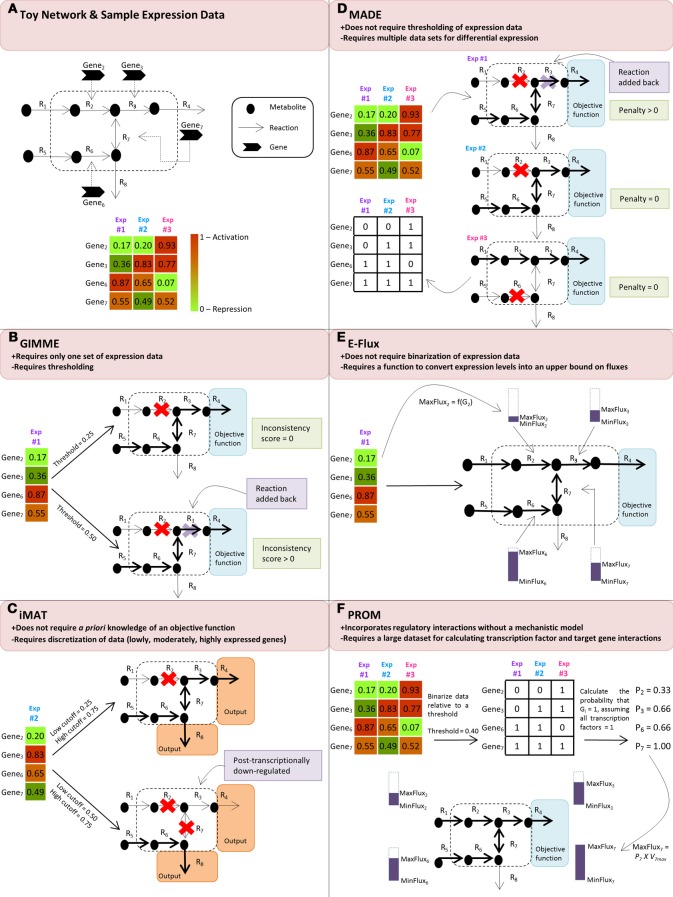
With the advent of high-throughput technologies, the field of systems biology has amassed an abundance of "omics" data, quantifying thousands of cellular components across a variety of scales, ranging from mRNA transcript levels to metabolite quantities. Methods are needed to not only integrate this omics data but to also use this data to heighten the predictive capabilities of computational models. Several recent studies have successfully demonstrated how flux balance analysis (FBA), a constraint-based modeling approach, can be used to integrate transcriptomic data into genome-scale metabolic network reconstructions to generate predictive computational models. In this review, we summarize such FBA-based methods for integrating expression data into genome-scale metabolic network reconstructions, highlighting their advantages as well as their limitations.
Keywords: data integration; expression data; flux balance analysis; metabolic networks; transcriptomics.
Figures
References
-
- Chang R. L., Ghamsari L., Manichaikul A., Hom E. F., Balaji S., Fu W., Shen Y., Hao T., Palsson B. O., Salehi-Ashtiani K., Papin J. A. (2011). Metabolic network reconstruction of Chlamydomonas offers insight into light-driven algal metabolism. Mol. Syst. Biol. 7, 1–13 10.1038/msb.2011.52 - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources