

Neuromuscular effects of G93A-SOD1 expression in zebrafish
- PMID: 22938571
- PMCID: PMC3506515
- DOI: 10.1186/1750-1326-7-44
Neuromuscular effects of G93A-SOD1 expression in zebrafish
Abstract
Background: Amyotrophic lateral sclerosis (ALS) is a fatal disorder involving the degeneration and loss of motor neurons. The mechanisms of motor neuron loss in ALS are unknown and there are no effective treatments. Defects in the distal axon and at the neuromuscular junction are early events in the disease course, and zebrafish provide a promising in vivo system to examine cellular mechanisms and treatments for these events in ALS pathogenesis.
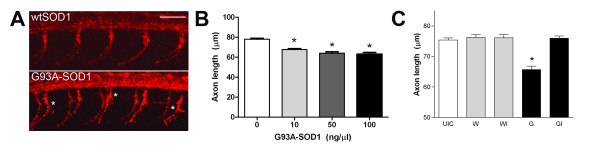
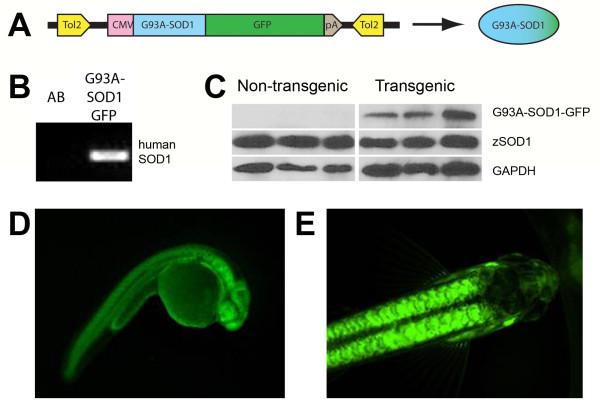
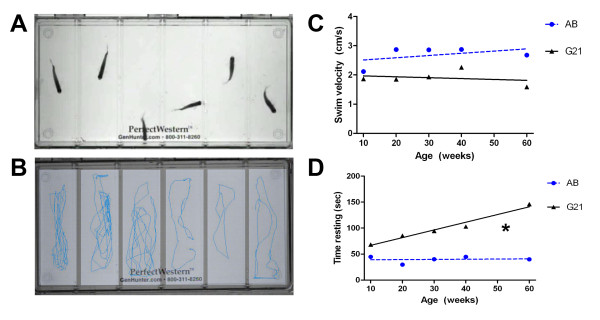
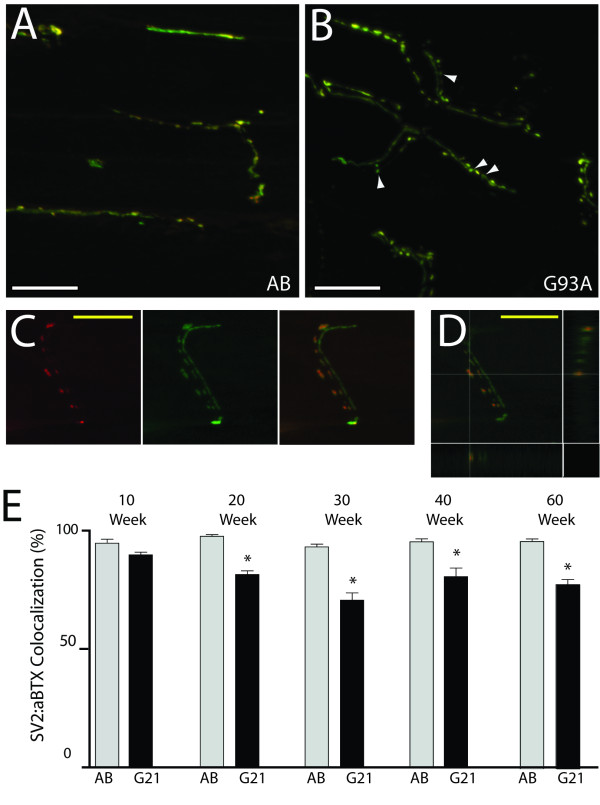
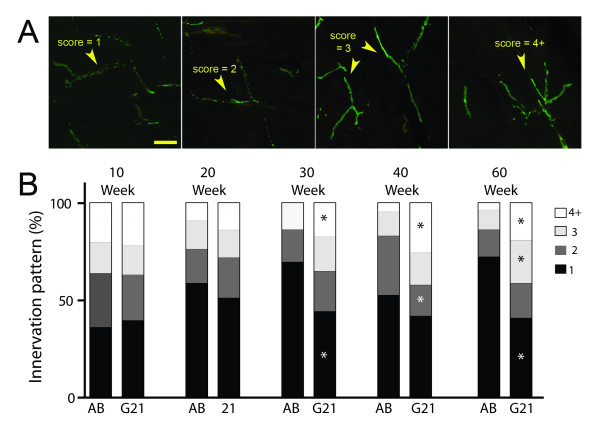
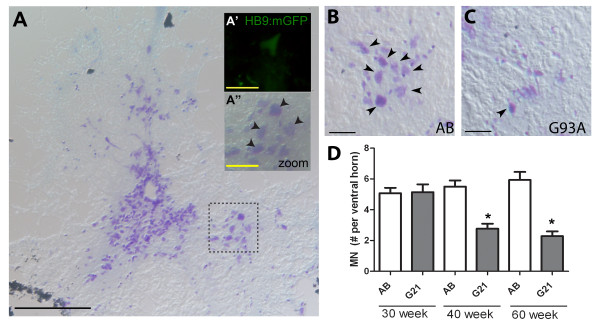
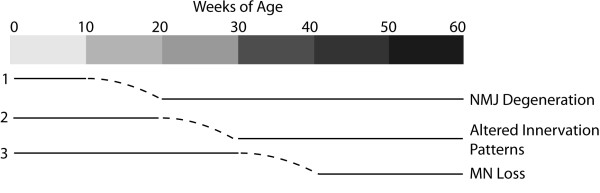
Results: We demonstrate that transient genetic manipulation of zebrafish to express G93A-SOD1, a mutation associated with familial ALS, results in early defects in motor neuron outgrowth and axonal branching. This is consistent with previous reports on motor neuron axonal defects associated with familial ALS genes following knockdown or mutant protein overexpression. We also demonstrate that upregulation of growth factor signaling is capable of rescuing these early defects, validating the potential of the model for therapeutic discovery. We generated stable transgenic zebrafish lines expressing G93A-SOD1 to further characterize the consequences of G93A-SOD1 expression on neuromuscular pathology and disease progression. Behavioral monitoring reveals evidence of motor dysfunction and decreased activity in transgenic ALS zebrafish. Examination of neuromuscular and neuronal pathology throughout the disease course reveals a loss of neuromuscular junctions and alterations in motor neuron innervations patterns with disease progression. Finally, motor neuron cell loss is evident later in the disease.
Conclusions: This sequence of events reflects the stepwise mechanisms of degeneration in ALS, and provides a novel model for mechanistic discovery and therapeutic development for neuromuscular degeneration in ALS.
Figures
Similar articles
-
Axonal degeneration, distal collateral branching and neuromuscular junction architecture alterations occur prior to symptom onset in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis.J Chem Neuroanat. 2016 Oct;76(Pt A):35-47. doi: 10.1016/j.jchemneu.2016.03.003. Epub 2016 Mar 30. J Chem Neuroanat. 2016. PMID: 27038603
-
AAV-NRIP gene therapy ameliorates motor neuron degeneration and muscle atrophy in ALS model mice.Skelet Muscle. 2024 Jul 24;14(1):17. doi: 10.1186/s13395-024-00349-z. Skelet Muscle. 2024. PMID: 39044305 Free PMC article.
-
Epothilone D accelerates disease progression in the SOD1G93A mouse model of amyotrophic lateral sclerosis.Neuropathol Appl Neurobiol. 2018 Oct;44(6):590-605. doi: 10.1111/nan.12473. Epub 2018 Mar 27. Neuropathol Appl Neurobiol. 2018. PMID: 29380402
-
Transgenic mice with human mutant genes causing Parkinson's disease and amyotrophic lateral sclerosis provide common insight into mechanisms of motor neuron selective vulnerability to degeneration.Rev Neurosci. 2007;18(2):115-36. doi: 10.1515/revneuro.2007.18.2.115. Rev Neurosci. 2007. PMID: 17593875 Review.
-
Neuromuscular junction destruction during amyotrophic lateral sclerosis: insights from transgenic models.Curr Opin Pharmacol. 2009 Jun;9(3):341-6. doi: 10.1016/j.coph.2009.03.007. Epub 2009 Apr 20. Curr Opin Pharmacol. 2009. PMID: 19386549 Review.
Cited by
-
Diving deep: zebrafish models in motor neuron degeneration research.Front Neurosci. 2024 Jun 20;18:1424025. doi: 10.3389/fnins.2024.1424025. eCollection 2024. Front Neurosci. 2024. PMID: 38966756 Free PMC article. Review.
-
Conditional Overexpression of rtn4al in Muscle of Adult Zebrafish Displays Defects Similar to Human Amyotrophic Lateral Sclerosis.Mar Biotechnol (NY). 2019 Feb;21(1):52-64. doi: 10.1007/s10126-018-9857-x. Epub 2018 Nov 15. Mar Biotechnol (NY). 2019. PMID: 30443836
-
Atomic structure of a toxic, oligomeric segment of SOD1 linked to amyotrophic lateral sclerosis (ALS).Proc Natl Acad Sci U S A. 2017 Aug 15;114(33):8770-8775. doi: 10.1073/pnas.1705091114. Epub 2017 Jul 31. Proc Natl Acad Sci U S A. 2017. PMID: 28760994 Free PMC article.
-
INaP selective inhibition reverts precocious inter- and motorneurons hyperexcitability in the Sod1-G93R zebrafish ALS model.Sci Rep. 2016 Apr 15;6:24515. doi: 10.1038/srep24515. Sci Rep. 2016. PMID: 27079797 Free PMC article.
-
Reduction of oxidative stress suppresses poly-GR-mediated toxicity in zebrafish embryos.Dis Model Mech. 2021 Nov 1;14(11):dmm049092. doi: 10.1242/dmm.049092. Epub 2021 Dec 1. Dis Model Mech. 2021. PMID: 34693978 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
