

Clinical approach to the diagnostic evaluation of hereditary and acquired neuromuscular diseases
- PMID: 22938875
- PMCID: PMC3482409
- DOI: 10.1016/j.pmr.2012.06.011
Clinical approach to the diagnostic evaluation of hereditary and acquired neuromuscular diseases
Abstract
For diagnostic evaluation of a neuromuscular disease, the clinician must be able to obtain a relevant patient and family history and perform focused general, musculoskeletal, neurologic, and functional physical examinations to direct further diagnostic evaluations. Laboratory studies for hereditary neuromuscular diseases include the relevant molecular genetic studies. The electromyogram and nerve-conduction studies remain an extension of the physical examination, and help to guide further diagnostic studies such as molecular genetics and muscle and nerve biopsies. All diagnostic information needs are to be interpreted within the context of relevant historical information, family history, physical examination, laboratory data, electrophysiology, pathology, and molecular genetics.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
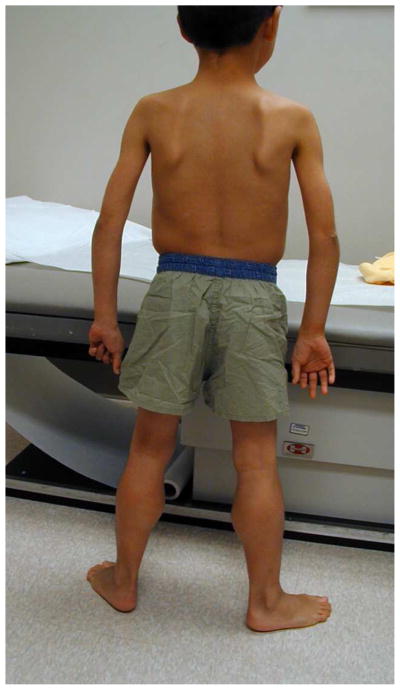
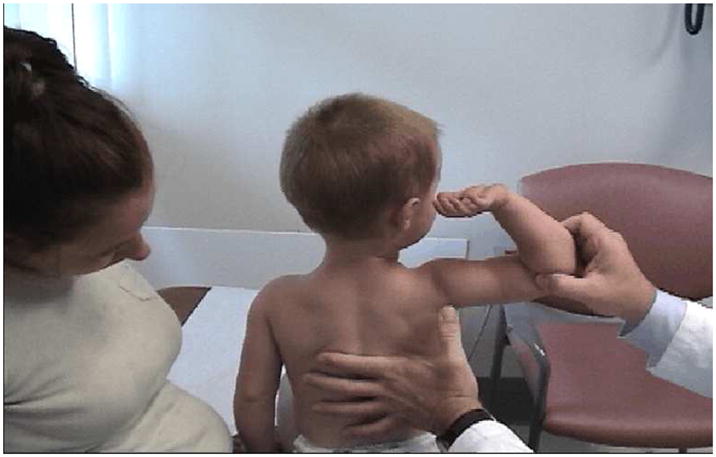
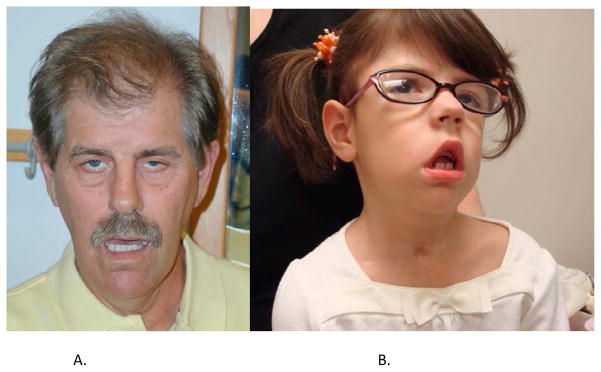
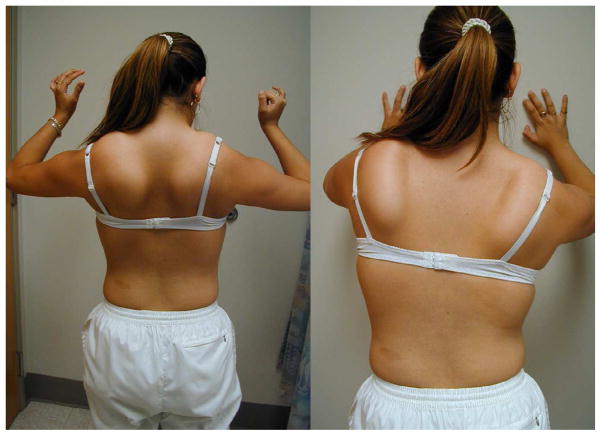



References
-
- Aitkens S, Lord J, Bernauer E, Fowler WM, Jr, Lieberman JS, Berck P. Relationship of manual muscle testing to objective strength measurements. Muscle Nerve. 1989;12:173–177. - PubMed
-
- Aitkens SG, McCrory MA, Kilmer DD, Bernauer EM. Moderate resistance exercise program: Its effect in slowly progressive neuromuscular disease. Arch Phys Med Rehabil. 1993;74:711–715. - PubMed
-
- Andersen PM, Nilsson P, Keranen M-L, et al. Phenotypic hetrogeneity in motor neuron disease patiens with CuZn-superoxide dismutase mutations in Scandanavia. Brain. 1997;120:1723–37. - PubMed
-
- Arkin AM. Absolute muscle power: The internal kinesiology of muscle, thesis. Department of Orthopedic Surgery. State University of Iowa; 1939.
-
- Arnold WD, Flanigan KM. A Practical Approach to Molecular Diagnostic Testing in Neuromuscular Diseases, Physical Medicine and Rehabilitation Clinics of North America. 2012. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
