

Using ERDS to infer copy-number variants in high-coverage genomes
- PMID: 22939633
- PMCID: PMC3511991
- DOI: 10.1016/j.ajhg.2012.07.004
Using ERDS to infer copy-number variants in high-coverage genomes
Abstract
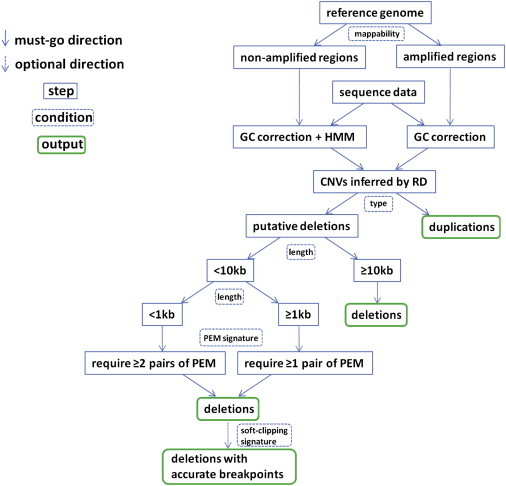
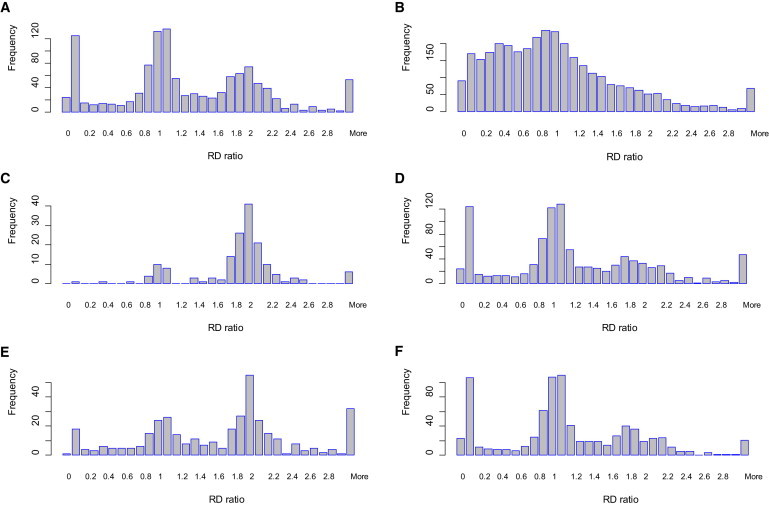
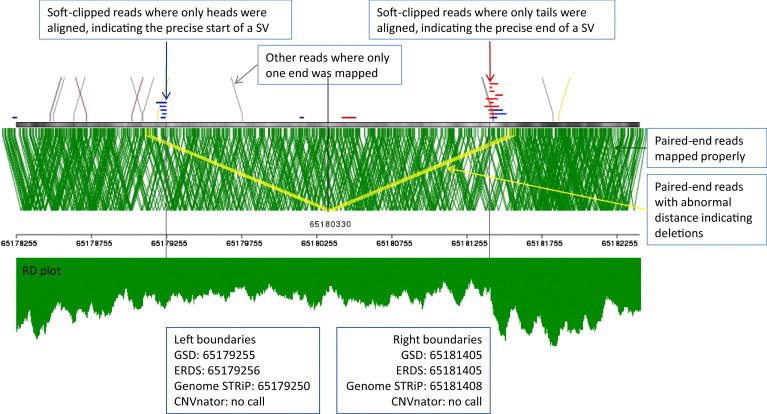
Although there are many methods available for inferring copy-number variants (CNVs) from next-generation sequence data, there remains a need for a system that is computationally efficient but that retains good sensitivity and specificity across all types of CNVs. Here, we introduce a new method, estimation by read depth with single-nucleotide variants (ERDS), and use various approaches to compare its performance to other methods. We found that for common CNVs and high-coverage genomes, ERDS performs as well as the best method currently available (Genome STRiP), whereas for rare CNVs and high-coverage genomes, ERDS performs better than any available method. Importantly, ERDS accommodates both unique and highly amplified regions of the genome and does so without requiring separate alignments for calling CNVs and other variants. These comparisons show that for genomes sequenced at high coverage, ERDS provides a computationally convenient method that calls CNVs as well as or better than any currently available method.
Copyright © 2012 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Sebat J., Lakshmi B., Troge J., Alexander J., Young J., Lundin P., Månér S., Massa H., Walker M., Chi M. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. - PubMed
-
- Iafrate A.J., Feuk L., Rivera M.N., Listewnik M.L., Donahoe P.K., Qi Y., Scherer S.W., Lee C. Detection of large-scale variation in the human genome. Nat. Genet. 2004;36:949–951. - PubMed
-
- Cook E.H., Jr., Scherer S.W. Copy-number variations associated with neuropsychiatric conditions. Nature. 2008;455:919–923. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
