

Endothelium-neutrophil interactions in ANCA-associated diseases
- PMID: 22942199
- PMCID: PMC3431419
- DOI: 10.1681/ASN.2012020119
Endothelium-neutrophil interactions in ANCA-associated diseases
Abstract
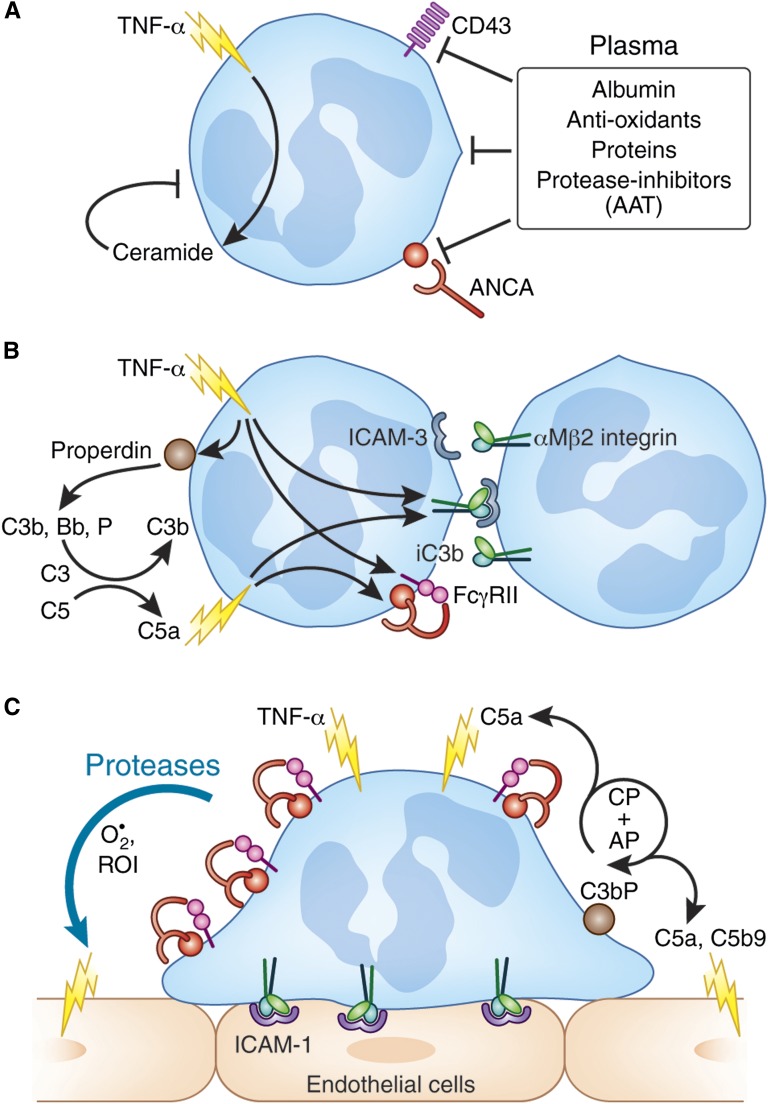
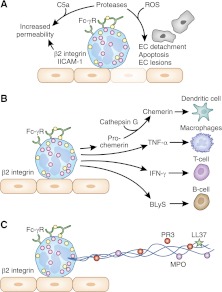
The two salient features of ANCA-associated vasculitis (AAV) are the restricted microvessel localization and the mechanism of inflammatory damage, independent of vascular immune deposits. The microvessel localization of the disease is due to the ANCA antigen accessibility, which is restricted to the membrane of neutrophils engaged in β2-integrin-mediated adhesion, while these antigens are cytoplasmic and inaccessible in resting neutrophils. The inflammatory vascular damage is the consequence of maximal proinflammatory responses of neutrophils, which face cumulative stimulations by TNF-α, β2-integrin engagement, C5a, and ANCA by the FcγRII receptor. This results in the premature intravascular explosive release by adherent neutrophils of all of their available weapons, normally designed to kill IgG-opsonized bacteria after migration in infected tissues.
Figures




References
-
- Wainwright J, Davson J: The renal appearances in the microscopic form of periarteritis nodosa. J Pathol Bacteriol 62: 189–196, 1950 - PubMed
-
- Jennette JC, Falk RJ, Andrassy K, Bacon PA, Churg J, Gross WL, Hagen EC, Hoffman GS, Hunder GG, Kallenberg CG, McCluskey RT, Sinico RA, Rees AJ, van Es LA, Waldherr R, Wiik A: Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum 37: 187–192, 1994 - PubMed
-
- Jennette JC, Falk RJ: Small-vessel vasculitis. N Engl J Med 337: 1512–1523, 1997 - PubMed
-
- van der Woude FJ, Rasmussen N, Lobatto S, Wiik A, Permin H, van Es LA, van der Giessen M, van der Hem GK, The TH: Autoantibodies against neutrophils and monocytes: Tool for diagnosis and marker of disease activity in Wegener’s granulomatosis. Lancet 1: 425–429, 1985 - PubMed
-
- Falk RJ, Jennette JC: Anti-neutrophil cytoplasmic autoantibodies with specificity for myeloperoxidase in patients with systemic vasculitis and idiopathic necrotizing and crescentic glomerulonephritis. N Engl J Med 318: 1651–1657, 1988 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
