

Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner
- PMID: 22942289
- PMCID: PMC3481266
- DOI: 10.1074/jbc.M112.393637
Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner
Abstract
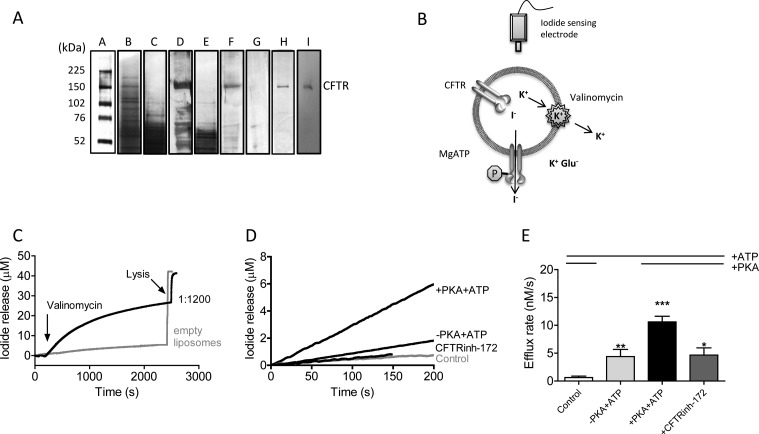
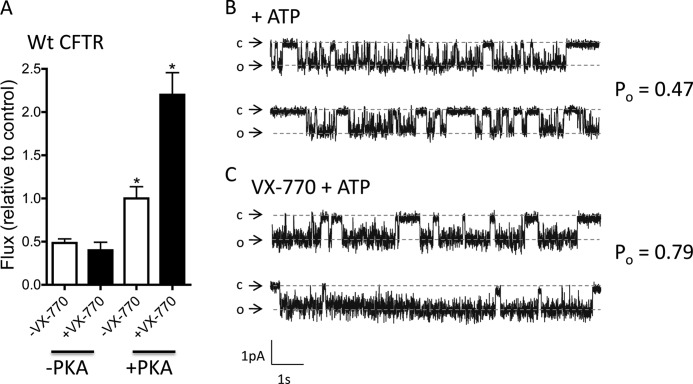
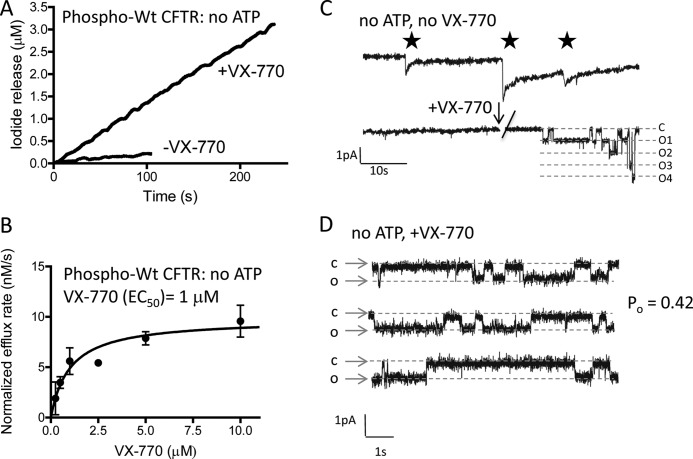
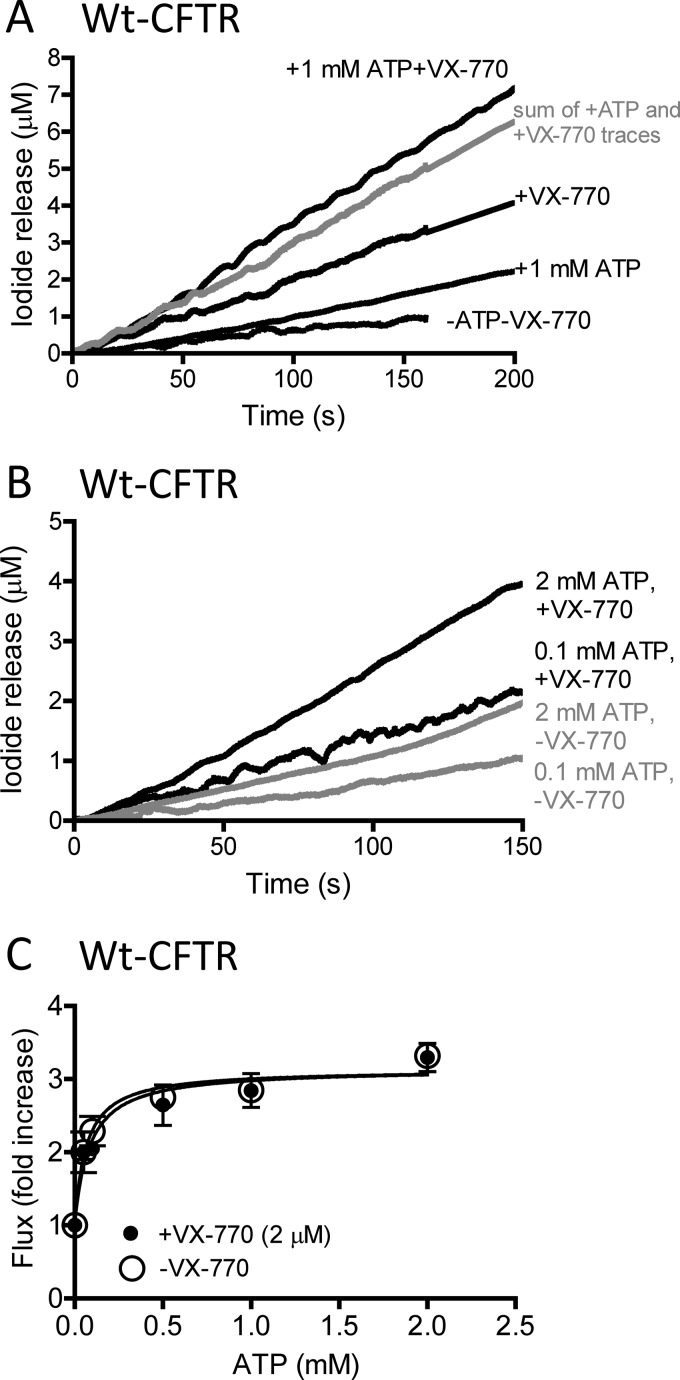
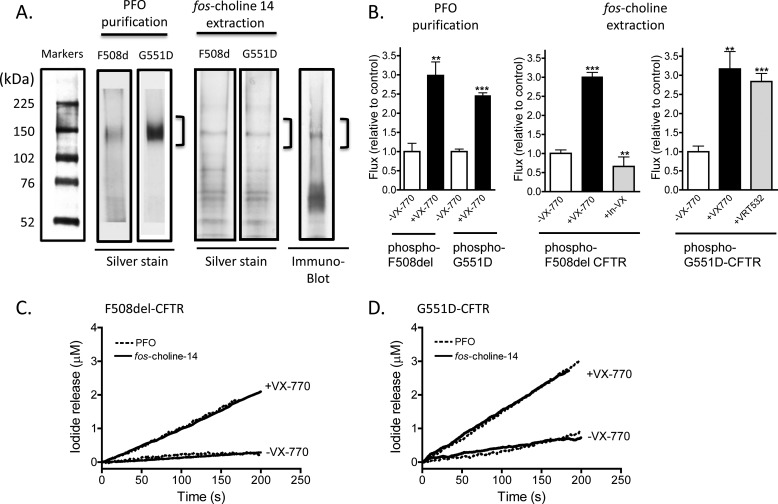
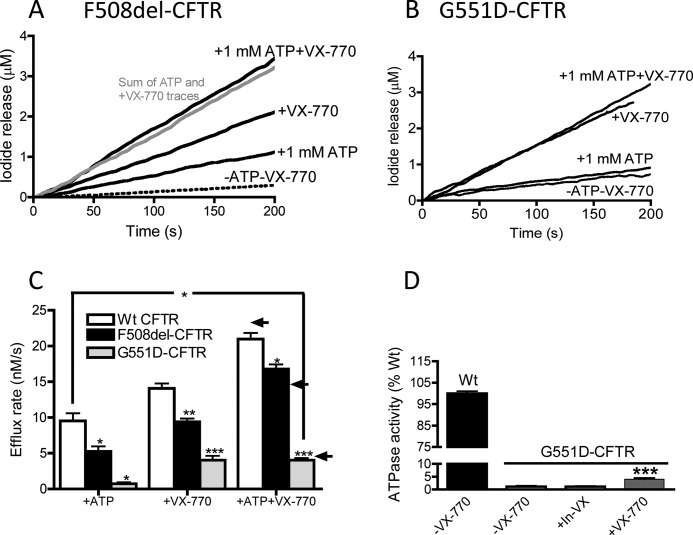
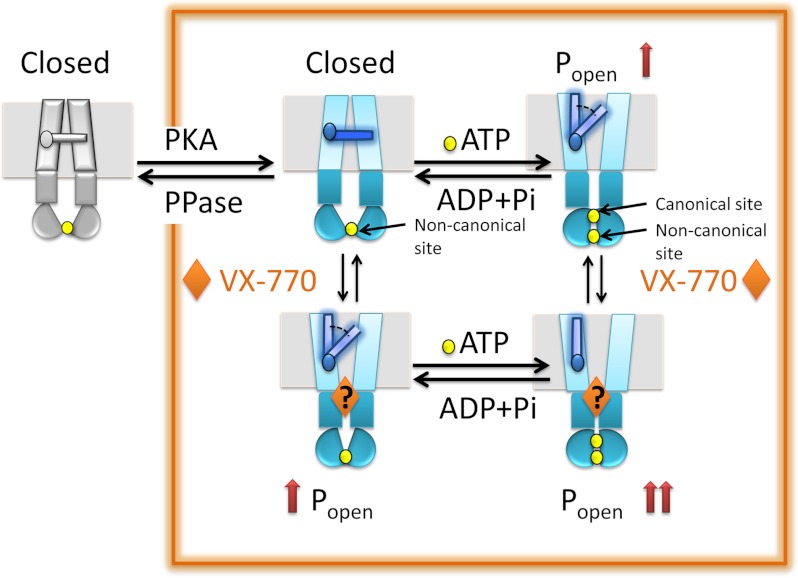
The cystic fibrosis transmembrane conductance regulator (CFTR) acts as a channel on the apical membrane of epithelia. Disease-causing mutations in the cystic fibrosis gene can lead to CFTR protein misfolding as in the case of the F508del mutation and/or channel dysfunction. Recently, a small molecule, VX-770 (ivacaftor), has shown efficacy in restoring lung function in patients bearing the G551D mutation, and this has been linked to repair of its channel gating defect. However, these studies did not reveal the mechanism of action of VX-770 in detail. Normally, CFTR channel activity is regulated by phosphorylation, ATP binding, and hydrolysis. Hence, it has been hypothesized that VX-770 modifies one or more of these metabolic events. In this study, we examined VX-770 activity using a reconstitution system for purified CFTR protein, a system that enables control of known regulatory factors. We studied the consequences of VX-770 interaction with CFTR incorporated in planar lipid bilayers and in proteoliposomes, using a novel flux-based assay. We found that purified and phosphorylated CFTR was potentiated in the presence of Mg-ATP, suggesting that VX-770 bound directly to the CFTR protein, rather than associated kinases or phosphatases. Interestingly, we also found that VX-770 enhanced the channel activity of purified and mutant CFTR in the nominal absence of Mg-ATP. These findings suggest that VX-770 can cause CFTR channel opening through a nonconventional ATP-independent mechanism. This work sets the stage for future studies of the structural properties that mediate CFTR gating using VX-770 as a probe.
Figures
References
-
- Proesmans M., Vermeulen F., De Boeck K. (2008) What's new in cystic fibrosis? From treating symptoms to correction of the basic defect. Eur. J. Pediatr. 167, 839–849 - PubMed
-
- Cheng S. H., Gregory R. J., Marshall J., Paul S., Souza D. W., White G. A., O'Riordan C. R., Smith A. E. (1990) Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63, 827–834 - PubMed
-
- Denning G. M., Anderson M. P., Amara J. F., Marshall J., Smith A. E., Welsh M. J. (1992) Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 358, 761–764 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
