

Mutations in FKBP10, which result in Bruck syndrome and recessive forms of osteogenesis imperfecta, inhibit the hydroxylation of telopeptide lysines in bone collagen
- PMID: 22949511
- PMCID: PMC3606010
- DOI: 10.1093/hmg/dds371
Mutations in FKBP10, which result in Bruck syndrome and recessive forms of osteogenesis imperfecta, inhibit the hydroxylation of telopeptide lysines in bone collagen
Abstract
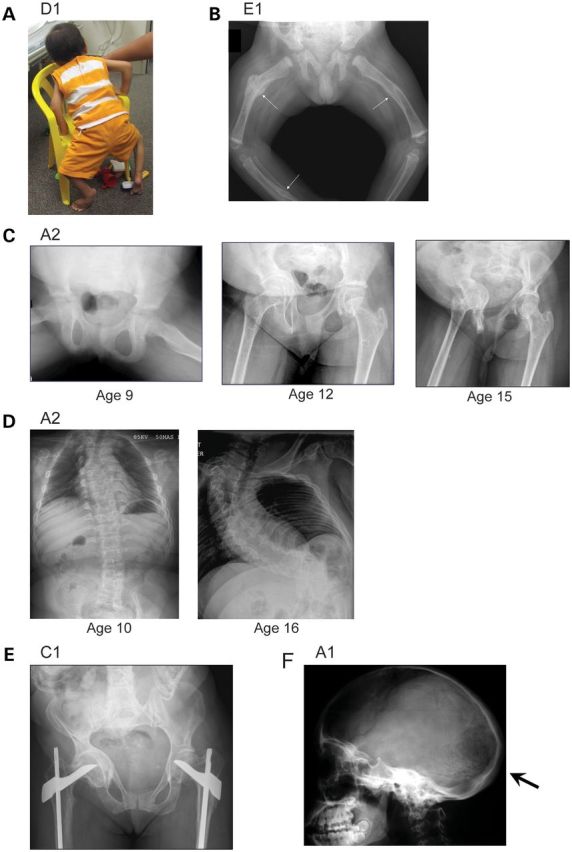
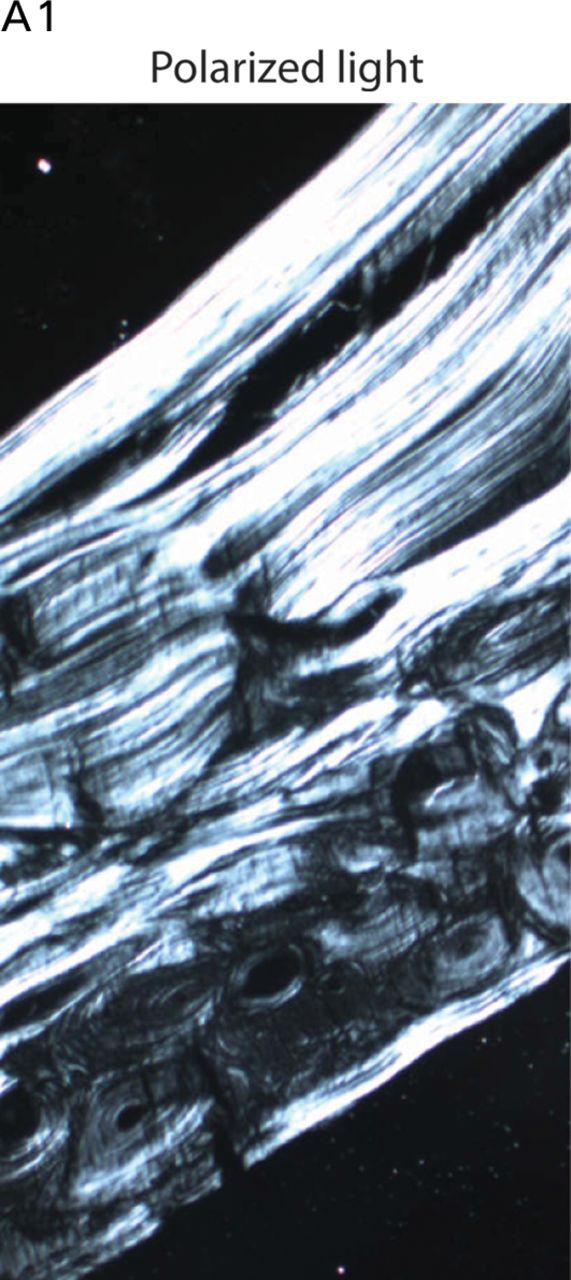
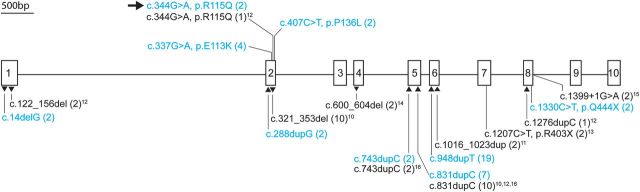
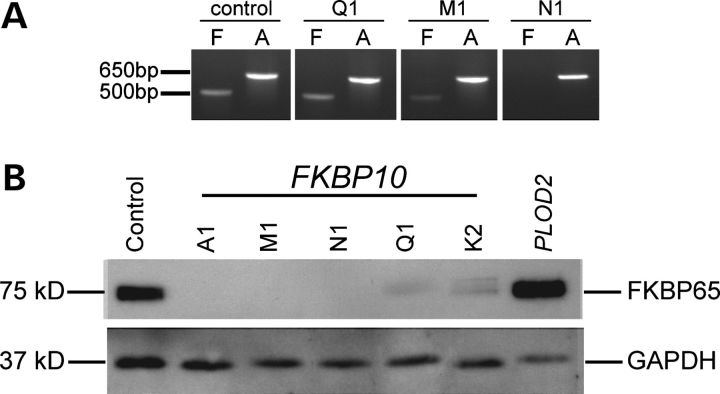
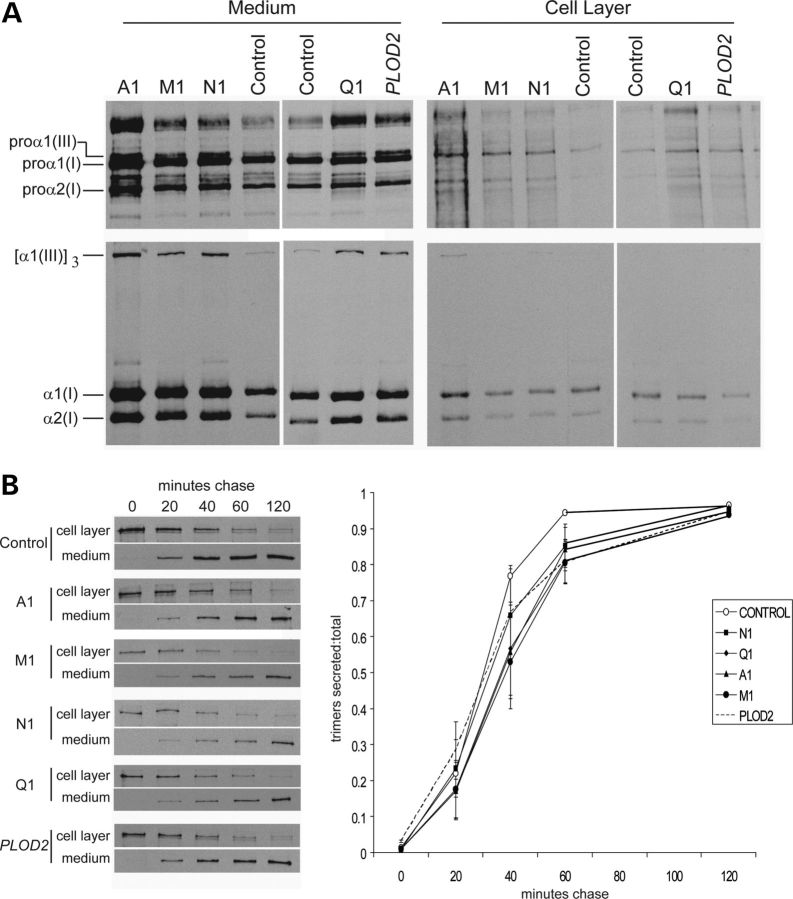
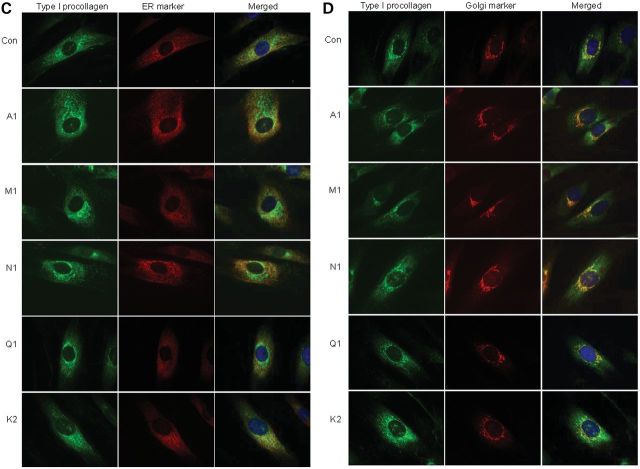
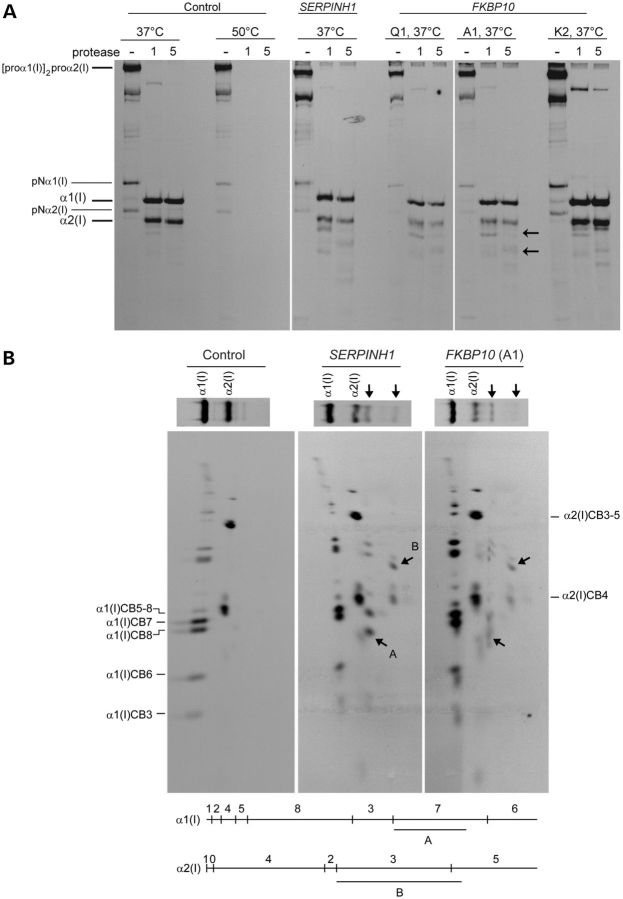
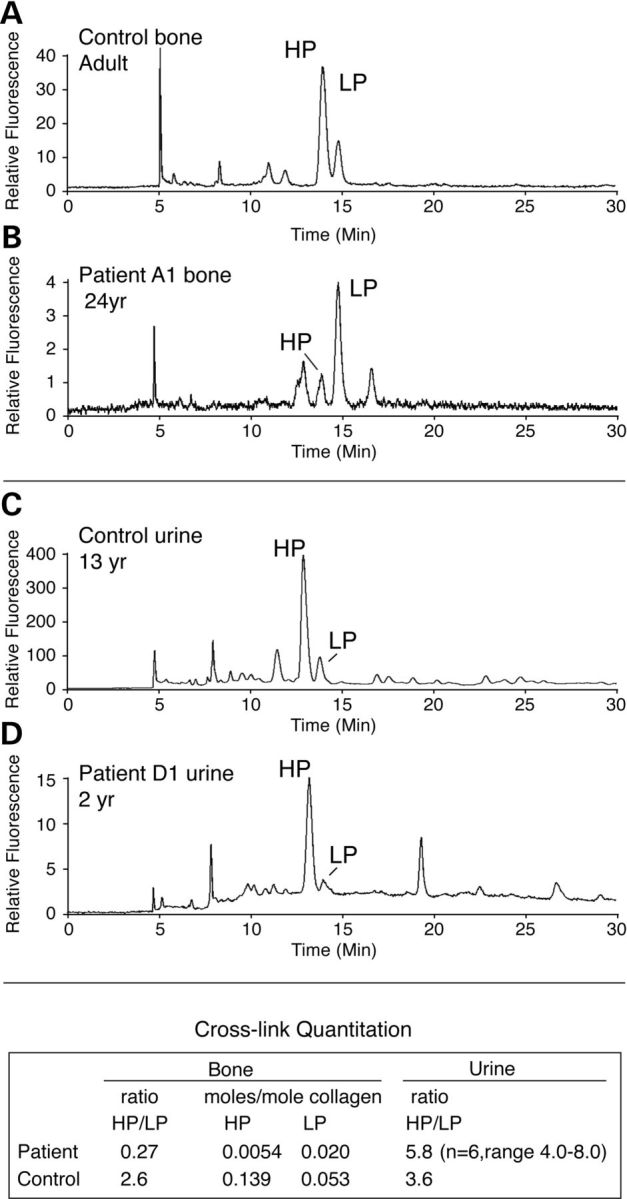
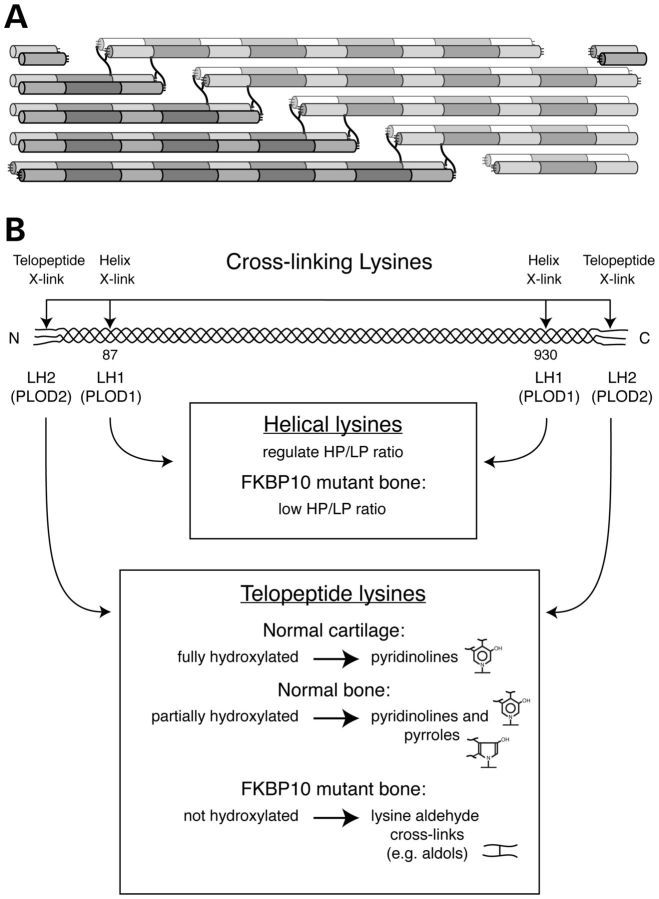
Although biallelic mutations in non-collagen genes account for <10% of individuals with osteogenesis imperfecta, the characterization of these genes has identified new pathways and potential interventions that could benefit even those with mutations in type I collagen genes. We identified mutations in FKBP10, which encodes the 65 kDa prolyl cis-trans isomerase, FKBP65, in 38 members of 21 families with OI. These include 10 families from the Samoan Islands who share a founder mutation. Of the mutations, three are missense; the remainder either introduce premature termination codons or create frameshifts both of which result in mRNA instability. In four families missense mutations result in loss of most of the protein. The clinical effects of these mutations are short stature, a high incidence of joint contractures at birth and progressive scoliosis and fractures, but there is remarkable variability in phenotype even within families. The loss of the activity of FKBP65 has several effects: type I procollagen secretion is slightly delayed, the stabilization of the intact trimer is incomplete and there is diminished hydroxylation of the telopeptide lysyl residues involved in intermolecular cross-link formation in bone. The phenotype overlaps with that seen with mutations in PLOD2 (Bruck syndrome II), which encodes LH2, the enzyme that hydroxylates the telopeptide lysyl residues. These findings define a set of genes, FKBP10, PLOD2 and SERPINH1, that act during procollagen maturation to contribute to molecular stability and post-translational modification of type I procollagen, without which bone mass and quality are abnormal and fractures and contractures result.
Figures
Similar articles
-
Mutations in FKBP10 cause recessive osteogenesis imperfecta and Bruck syndrome.J Bone Miner Res. 2011 Mar;26(3):666-72. doi: 10.1002/jbmr.250. J Bone Miner Res. 2011. PMID: 20839288 Free PMC article.
-
Loss of the long form of Plod2 phenocopies contractures of Bruck syndrome-osteogenesis imperfecta.J Bone Miner Res. 2024 Sep 2;39(9):1240-1252. doi: 10.1093/jbmr/zjae124. J Bone Miner Res. 2024. PMID: 39088537 Free PMC article.
-
Kuskokwim syndrome, a recessive congenital contracture disorder, extends the phenotype of FKBP10 mutations.Hum Mutat. 2013 Sep;34(9):1279-88. doi: 10.1002/humu.22362. Epub 2013 Jul 8. Hum Mutat. 2013. PMID: 23712425 Free PMC article.
-
Bone collagen: new clues to its mineralization mechanism from recessive osteogenesis imperfecta.Calcif Tissue Int. 2013 Oct;93(4):338-47. doi: 10.1007/s00223-013-9723-9. Epub 2013 Mar 19. Calcif Tissue Int. 2013. PMID: 23508630 Free PMC article. Review.
-
[Mutations of noncollagen genes in osteogenesis imperfecta--implications of the gene products in collagen biosynthesis and pathogenesis of disease].Postepy Hig Med Dosw (Online). 2012 Jun 14;66:359-71. doi: 10.5604/17322693.1000336. Postepy Hig Med Dosw (Online). 2012. PMID: 22706122 Review. Polish.
Cited by
-
Exercise increases pyridinoline cross-linking and counters the mechanical effects of concurrent lathyrogenic treatment.Bone. 2015 Dec;81:327-337. doi: 10.1016/j.bone.2015.07.030. Epub 2015 Jul 23. Bone. 2015. PMID: 26211995 Free PMC article.
-
Arthrogryposis multiplex congenita in a child with congenital fractures: a case report.J Med Case Rep. 2022 Oct 19;16(1):376. doi: 10.1186/s13256-022-03587-1. J Med Case Rep. 2022. PMID: 36258204 Free PMC article.
-
Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment.Am J Med Genet A. 2014 Jun;164A(6):1470-81. doi: 10.1002/ajmg.a.36545. Epub 2014 Apr 8. Am J Med Genet A. 2014. PMID: 24715559 Free PMC article. Review.
-
Features of Congenital Arthrogryposis Due to Abnormalities in Collagen Homeostasis, a Scoping Review.Int J Mol Sci. 2023 Aug 31;24(17):13545. doi: 10.3390/ijms241713545. Int J Mol Sci. 2023. PMID: 37686358 Free PMC article.
-
Osteogenesis imperfecta due to mutations in non-collagenous genes: lessons in the biology of bone formation.Curr Opin Pediatr. 2014 Aug;26(4):500-7. doi: 10.1097/MOP.0000000000000117. Curr Opin Pediatr. 2014. PMID: 25007323 Free PMC article. Review.
References
-
- Morello R., Bertin T., Chen Y., Hicks J., Tonachini L., Monticone M., Castagnola P., Rauch F., Glorieux F., Vranka J., et al. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006;127:291–304. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
