

Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110α (PIK3CA)
- PMID: 22949682
- PMCID: PMC3458343
- DOI: 10.1073/pnas.1205508109
Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110α (PIK3CA)
Abstract
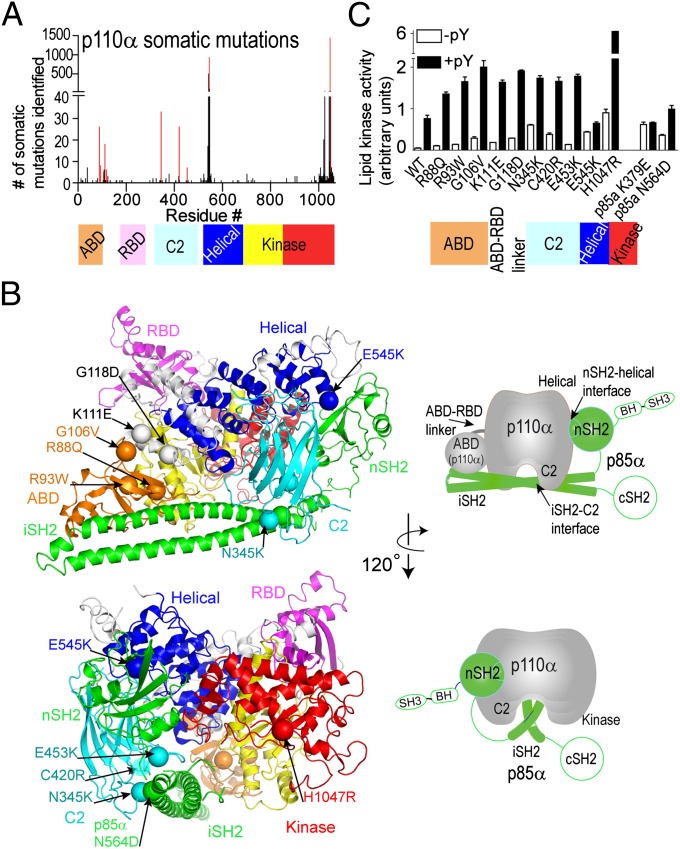
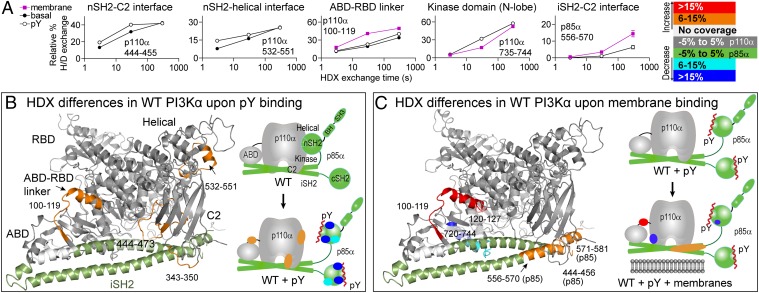
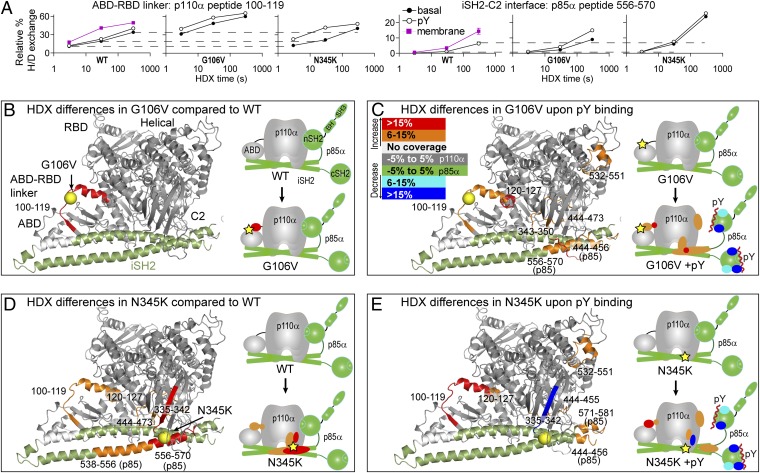
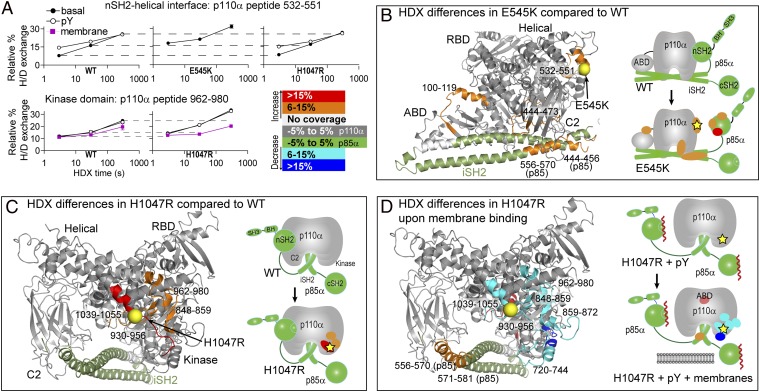
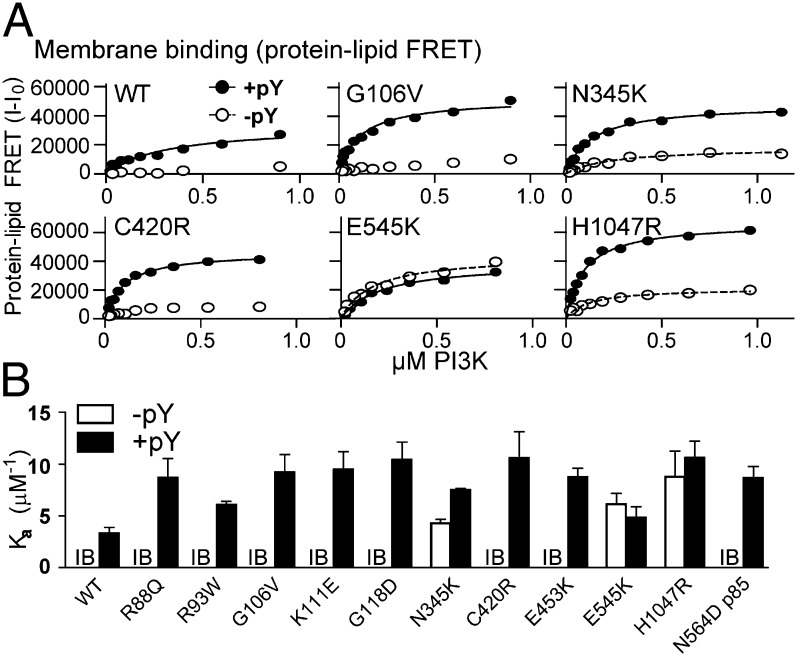
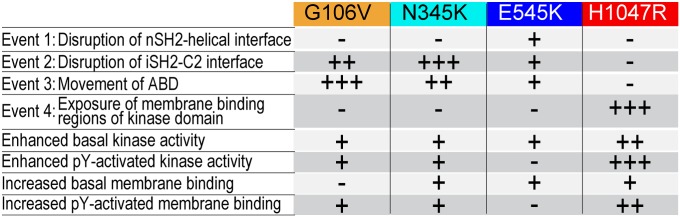
The p110α catalytic subunit (PIK3CA) is one of the most frequently mutated genes in cancer. We have examined the activation of the wild-type p110α/p85α and a spectrum of oncogenic mutants using hydrogen/deuterium exchange mass spectrometry (HDX-MS). We find that for the wild-type enzyme, the natural transition from an inactive cytosolic conformation to an activated form on membranes entails four distinct events. Analysis of oncogenic mutations shows that all up-regulate the enzyme by enhancing one or more of these dynamic events. We provide the first insight into the activation mechanism by mutations in the linker between the adapter-binding domain (ABD) and the Ras-binding domain (RBD) (G106V and G118D). These mutations, which are common in endometrial cancers, enhance two of the natural activation events: movement of the ABD and ABD-RBD linker relative to the rest of the catalytic subunit and breaking the C2-iSH2 interface on binding membranes. C2 domain mutants (N345K and C420R) also mimic these events, even in the absence of membranes. A third event is breaking the nSH2-helical domain contact caused by phosphotyrosine-containing peptides binding to the enzyme, which is mimicked by a helical domain mutation (E545K). Interaction of the C lobe of the kinase domain with membranes is the fourth activation event, and is potentiated by kinase domain mutations (e.g., H1047R). All mutations increased lipid binding and basal activity, even mutants distant from the membrane surface. Our results elucidate a unifying mechanism in which diverse PIK3CA mutations stimulate lipid kinase activity by facilitating allosteric motions required for catalysis on membranes.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Chang HW, et al. Transformation of chicken cells by the gene encoding the catalytic subunit of PI 3-kinase. Science. 1997;276:1848–1850. - PubMed
-
- Samuels Y, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. - PubMed
-
- Vadas O, Burke JE, Zhang X, Berndt A, Williams RL. Structural basis for activation and inhibition of class I phosphoinositide 3-kinases. Sci Signal. 2011;4:re2. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
