

Classification of human genomic regions based on experimentally determined binding sites of more than 100 transcription-related factors
- PMID: 22950945
- PMCID: PMC3491392
- DOI: 10.1186/gb-2012-13-9-r48
Classification of human genomic regions based on experimentally determined binding sites of more than 100 transcription-related factors
Abstract
Background: Transcription factors function by binding different classes of regulatory elements. The Encyclopedia of DNA Elements (ENCODE) project has recently produced binding data for more than 100 transcription factors from about 500 ChIP-seq experiments in multiple cell types. While this large amount of data creates a valuable resource, it is nonetheless overwhelmingly complex and simultaneously incomplete since it covers only a small fraction of all human transcription factors.
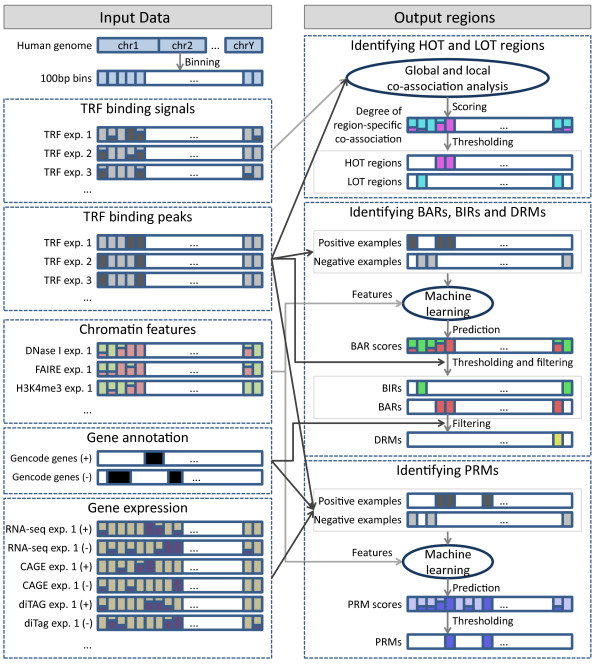
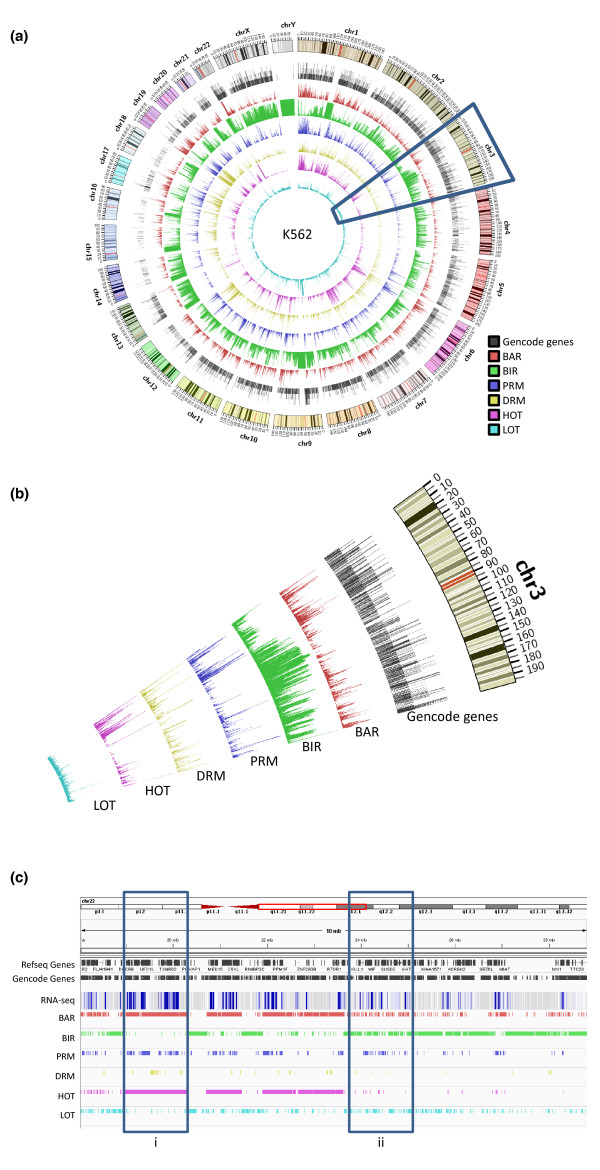
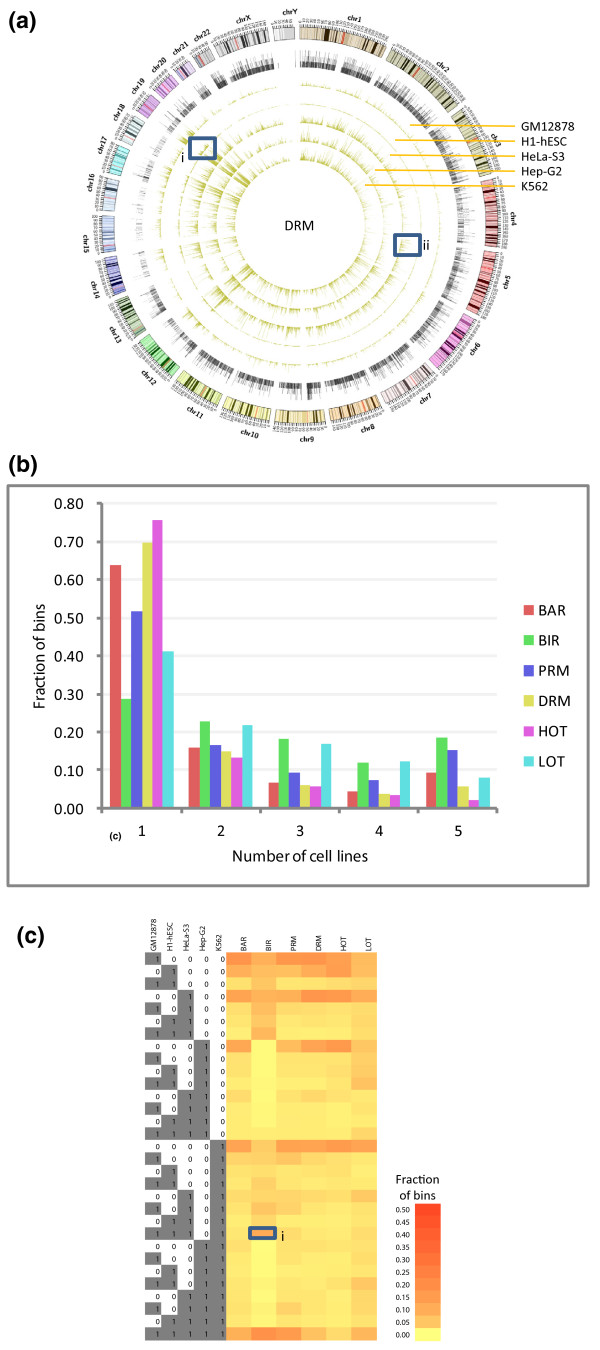
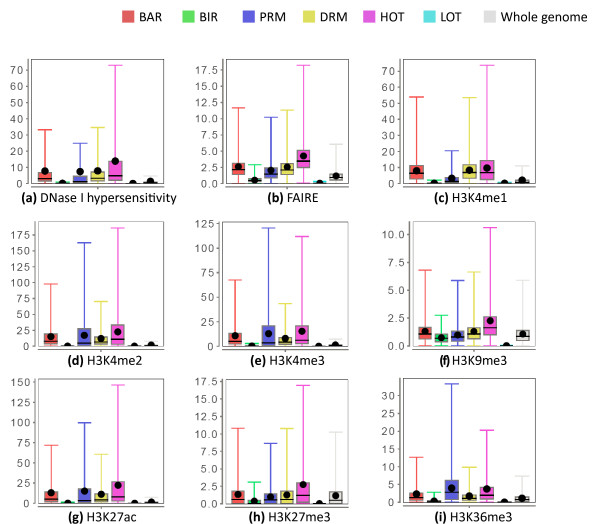
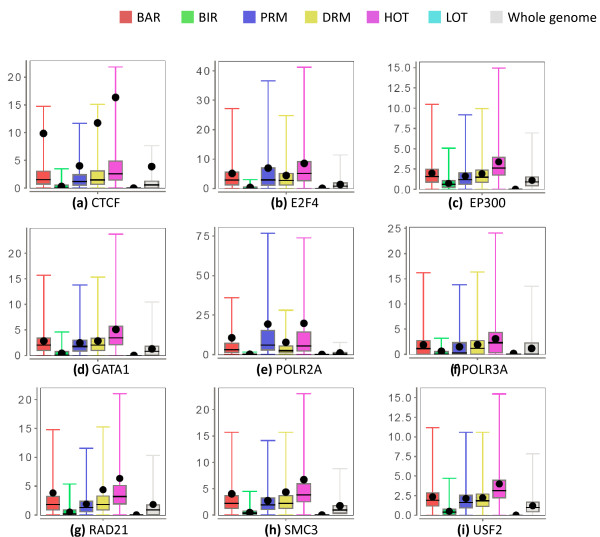
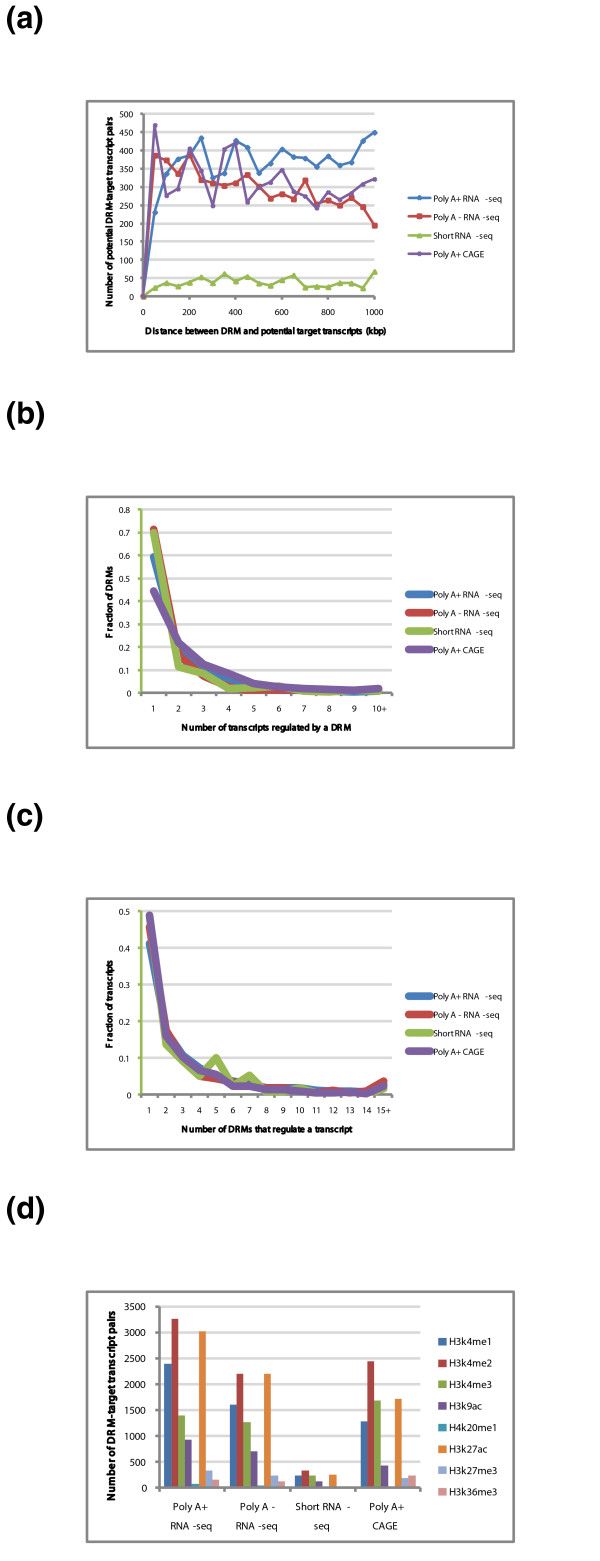
Results: As part of the consortium effort in providing a concise abstraction of the data for facilitating various types of downstream analyses, we constructed statistical models that capture the genomic features of three paired types of regions by machine-learning methods: firstly, regions with active or inactive binding; secondly, those with extremely high or low degrees of co-binding, termed HOT and LOT regions; and finally, regulatory modules proximal or distal to genes. From the distal regulatory modules, we developed computational pipelines to identify potential enhancers, many of which were validated experimentally. We further associated the predicted enhancers with potential target transcripts and the transcription factors involved. For HOT regions, we found a significant fraction of transcription factor binding without clear sequence motifs and showed that this observation could be related to strong DNA accessibility of these regions.
Conclusions: Overall, the three pairs of regions exhibit intricate differences in chromosomal locations, chromatin features, factors that bind them, and cell-type specificity. Our machine learning approach enables us to identify features potentially general to all transcription factors, including those not included in the data.
Figures
References
-
- Blackwood EM, Kadonaga JT. Going the distance: a current view of enhancer action. Science. 1998;281:60–63. - PubMed
