

Endoplasmic reticulum stress, pancreatic β-cell degeneration, and diabetes
- PMID: 22951443
- PMCID: PMC3426819
- DOI: 10.1101/cshperspect.a007666
Endoplasmic reticulum stress, pancreatic β-cell degeneration, and diabetes
Abstract
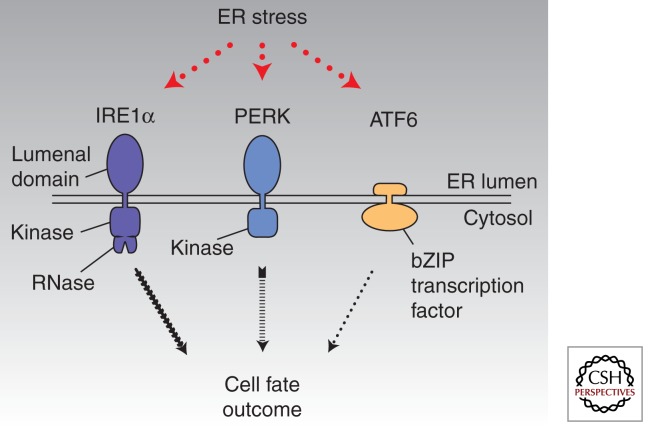
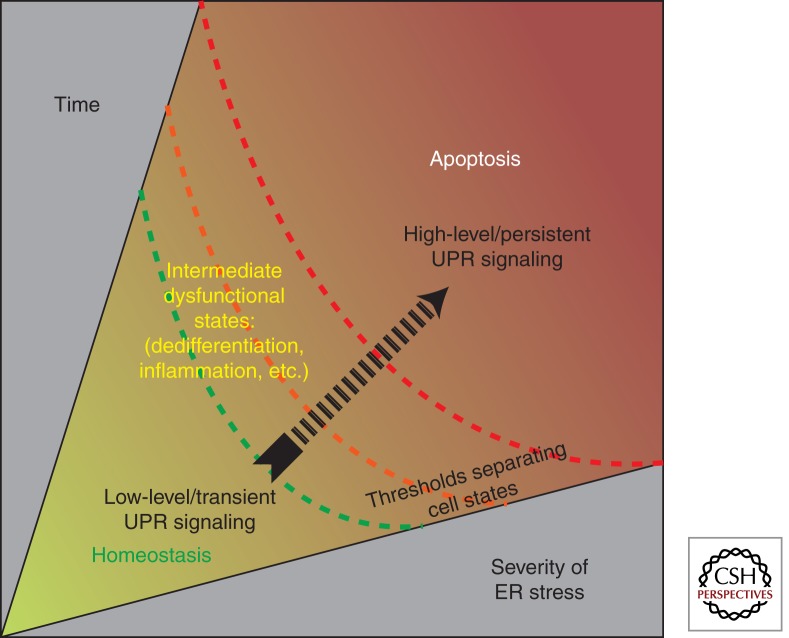
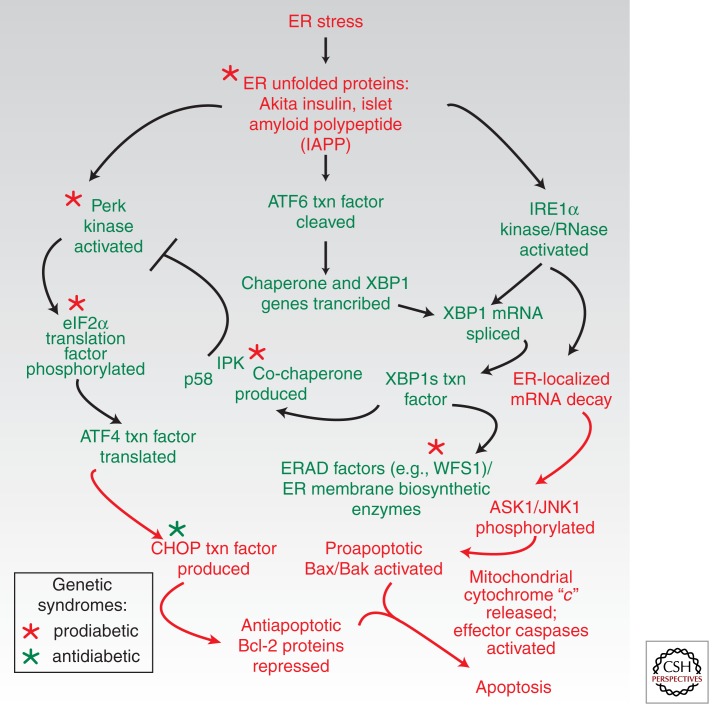
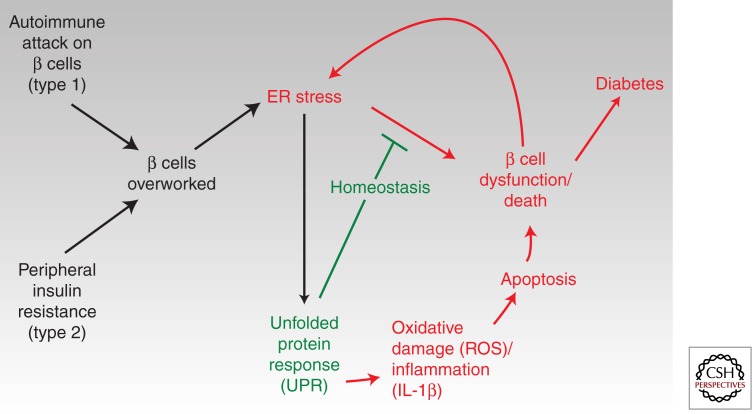
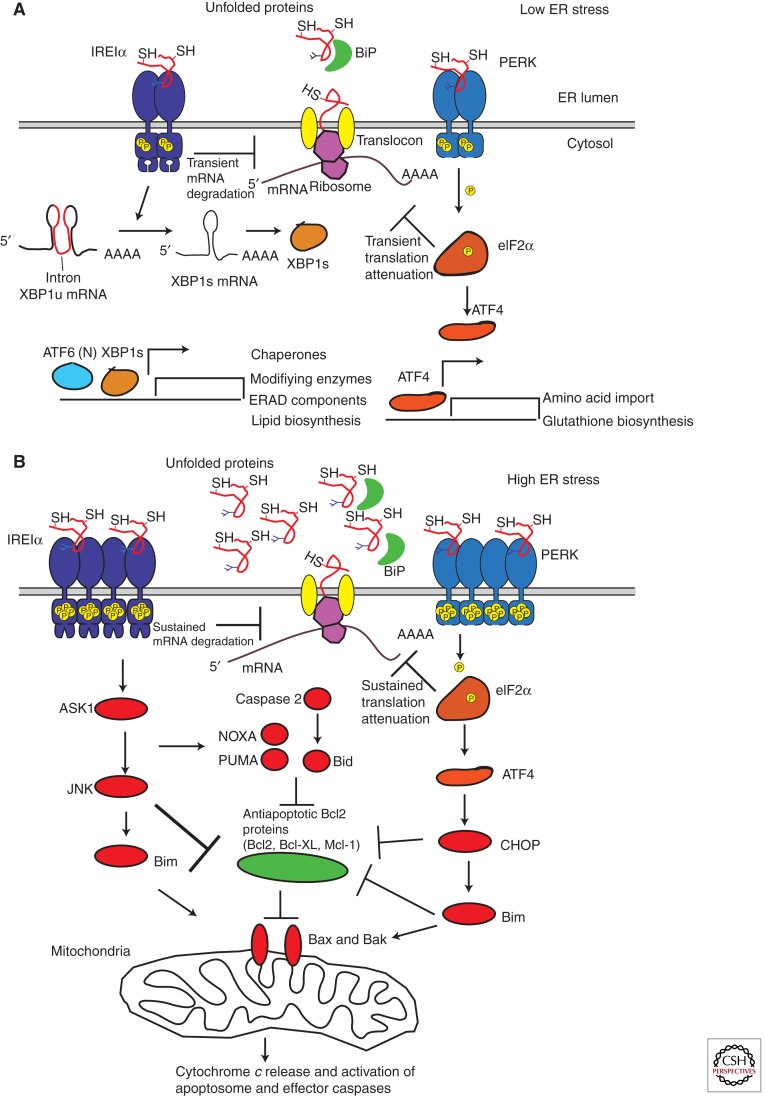
Overwhelming of protein folding in the endoplasmic reticulum (ER)--referred to as "ER stress"--activates a set of intracellular signaling pathways termed the unfolded protein response (UPR). Beneficial outputs of the UPR promote adaptation in cells experiencing manageably low levels of ER stress. However, if ER stress reaches critically high levels, the UPR uses destructive outputs to trigger programmed cell death. Genetic mutations in various UPR components cause inherited syndromes of diabetes mellitus in both rodents and humans, implicating the UPR in the proper functioning and survival of pancreatic islet β cells. Markers of chronically elevated ER stress, terminal UPR signaling, and apoptosis are evident in β cells in these rare disorders; these markers are similarly present in islets of human patients with common forms of diabetes. These findings promise to enhance our molecular understanding of human diabetes significantly and may lead to new and effective therapies.
Figures
References
-
- Bernales S, Papa FR, Walter P 2006. Intracellular signaling by the unfolded protein response. Annu Rev Cell Dev Biol 22: 487–508 - PubMed
-
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D 2000. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2: 326–332 - PubMed
-
- Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, et al. 2005. A selective inhibitor of eIF2α dephosphorylation protects cells from ER stress. Science 307: 935–939 - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical