

CFTR, mucins, and mucus obstruction in cystic fibrosis
- PMID: 22951447
- PMCID: PMC3426818
- DOI: 10.1101/cshperspect.a009589
CFTR, mucins, and mucus obstruction in cystic fibrosis
Abstract
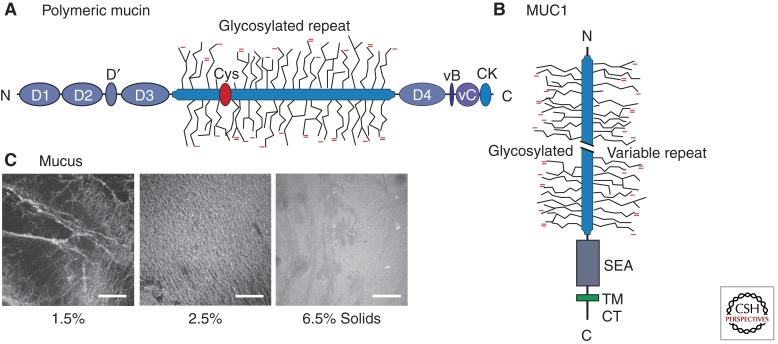
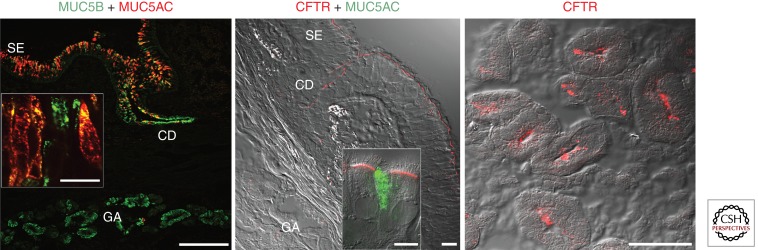
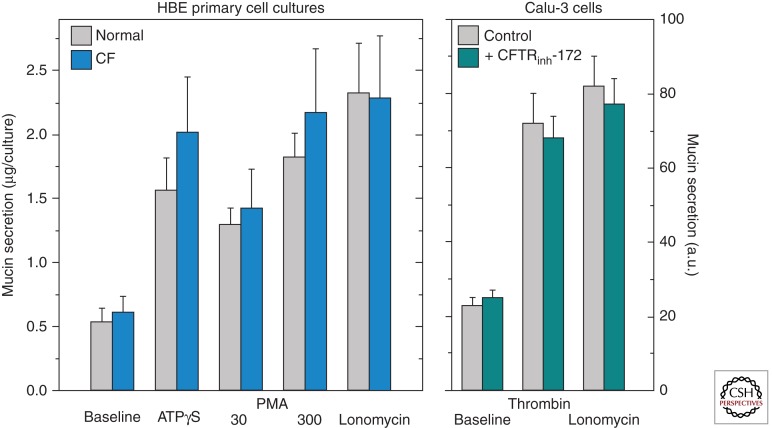
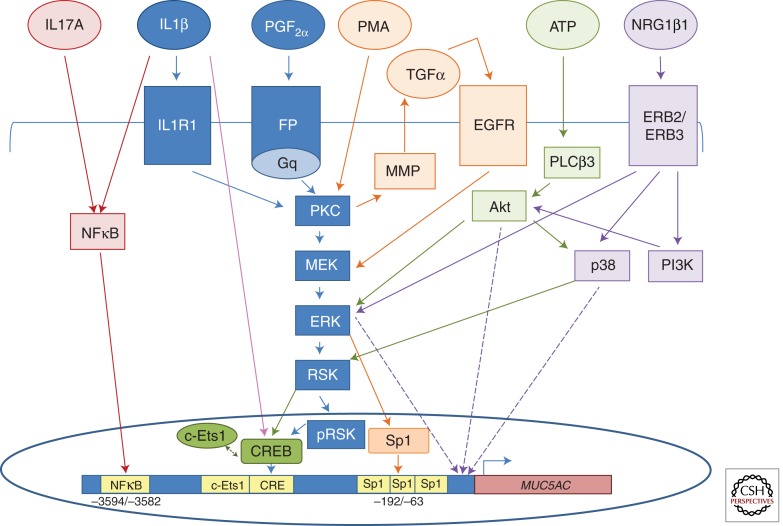
Mucus pathology in cystic fibrosis (CF) has been known for as long as the disease has been recognized and is sometimes called mucoviscidosis. The disease is marked by mucus hyperproduction and plugging in many organs, which are usually most fatal in the airways of CF patients, once the problem of meconium ileus at birth is resolved. After the CF gene, CFTR, was cloned and its protein product identified as a cAMP-regulated Cl(-) channel, causal mechanisms underlying the strong mucus phenotype of the disease became obscure. Here we focus on mucin genes and polymeric mucin glycoproteins, examining their regulation and potential relationships to a dysfunctional cystic fibrosis transmembrane conductance regulator (CFTR). Detailed examination of CFTR expression in organs and different cell types indicates that changes in CFTR expression do not always correlate with the severity of CF disease or mucus accumulation. Thus, the mucus hyperproduction that typifies CF does not appear to be a direct cause of a defective CFTR but, rather, to be a downstream consequence. In organs like the lung, up-regulation of mucin gene expression by inflammation results from chronic infection; however, in other instances and organs, the inflammation may have a non-infectious origin. The mucus plugging phenotype of the β-subunit of the epithelial Na(+) channel (βENaC)-overexpressing mouse is proving to be an archetypal example of this kind of inflammation, with a dehydrated airway surface/concentrated mucus gel apparently providing the inflammatory stimulus. Data indicate that the luminal HCO(3)(-) deficiency recently described for CF epithelia may also provide such a stimulus, perhaps by causing a mal-maturation of mucins as they are released onto luminal surfaces. In any event, the path between CFTR dysfunction and mucus hyperproduction has proven tortuous, and its unraveling continues to offer its own twists and turns, along with fascinating glimpses into biology.
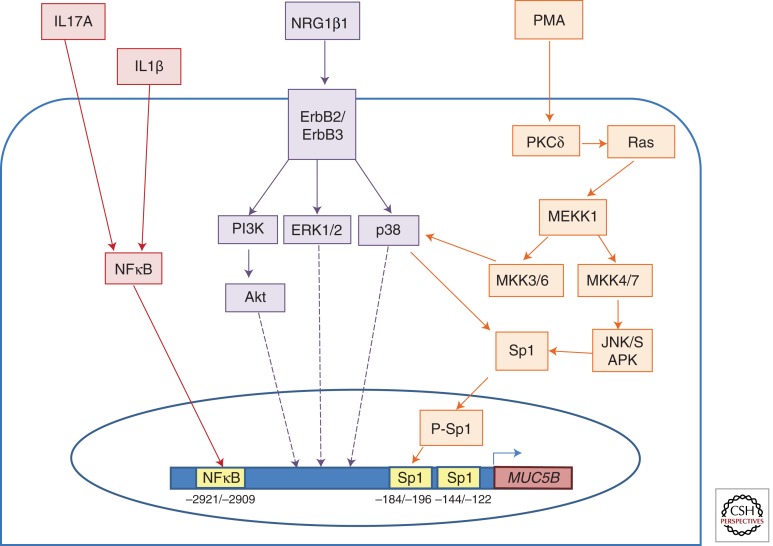
Figures
References
-
- Abdullah LH, Conway JD, Cohn JA, Davis CW 1997. Protein kinase C and Ca2+ activation of mucin secretion in airway goblet cells. Am J Physiol 273: L201–L210 - PubMed
-
- Ajonuma LC, Ng EH, Chow PH, Hung CY, Tsang LL, Cheung AN, Brito-Jones C, Lok IH, Haines J, Chan HC 2005. Increased cystic fibrosis transmembrane conductance regulator (CFTR) expression in the human hydrosalpinx. Hum Reprod 20: 1228–1234 - PubMed
-
- Allen A, Hutton DA, Pearson JP, Sellers LA 1984. Mucus glycoprotein structure, gel formation and gastrointestinal mucus function. Ciba Found Symp 109: 137–156 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P50 HL060280/HL/NHLBI NIH HHS/United States
- R01 HL063756/HL/NHLBI NIH HHS/United States
- HL063756/HL/NHLBI NIH HHS/United States
- R01 HL033152/HL/NHLBI NIH HHS/United States
- HL34322/HL/NHLBI NIH HHS/United States
- AI087717/AI/NIAID NIH HHS/United States
- HL 97000/HL/NHLBI NIH HHS/United States
- HL60280/HL/NHLBI NIH HHS/United States
- R01 HL097000/HL/NHLBI NIH HHS/United States
- P01 HL051818/HL/NHLBI NIH HHS/United States
- HL51818/HL/NHLBI NIH HHS/United States
- HL34582/HL/NHLBI NIH HHS/United States
- P01 HL034322/HL/NHLBI NIH HHS/United States
- R21 AI087717/AI/NIAID NIH HHS/United States
- HL 33152/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials