

Structures of human DPP7 reveal the molecular basis of specific inhibition and the architectural diversity of proline-specific peptidases
- PMID: 22952628
- PMCID: PMC3430648
- DOI: 10.1371/journal.pone.0043019
Structures of human DPP7 reveal the molecular basis of specific inhibition and the architectural diversity of proline-specific peptidases
Abstract
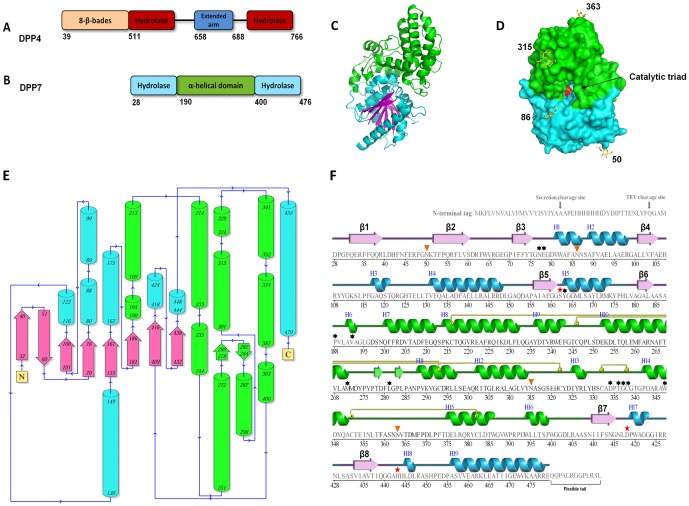
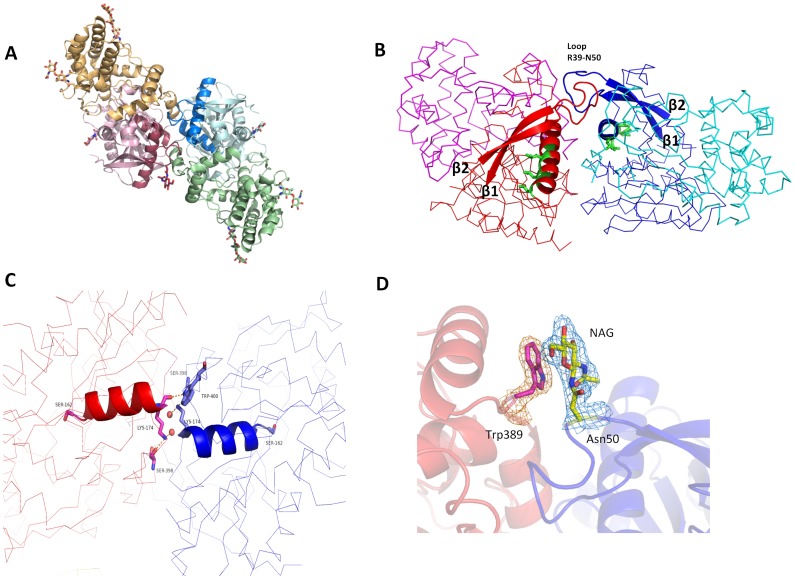
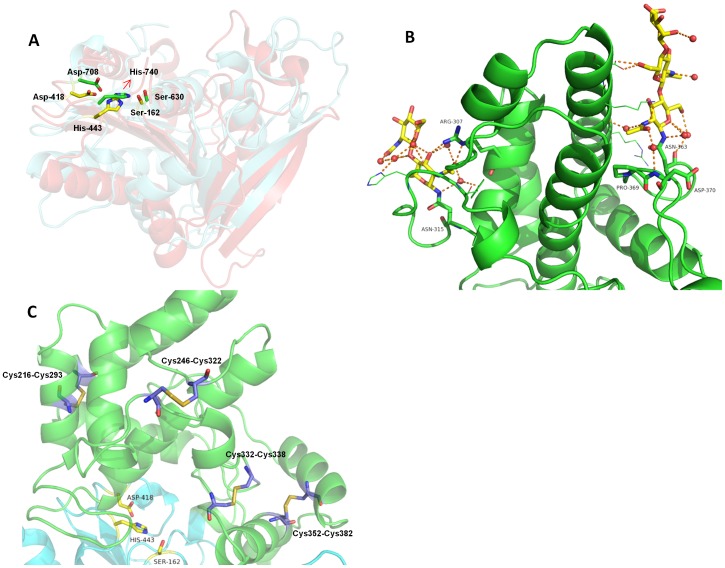
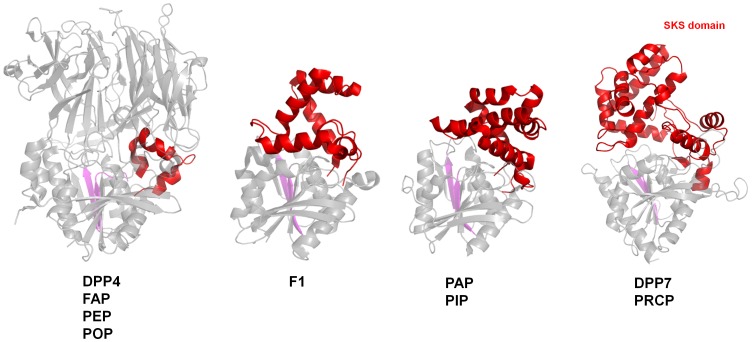
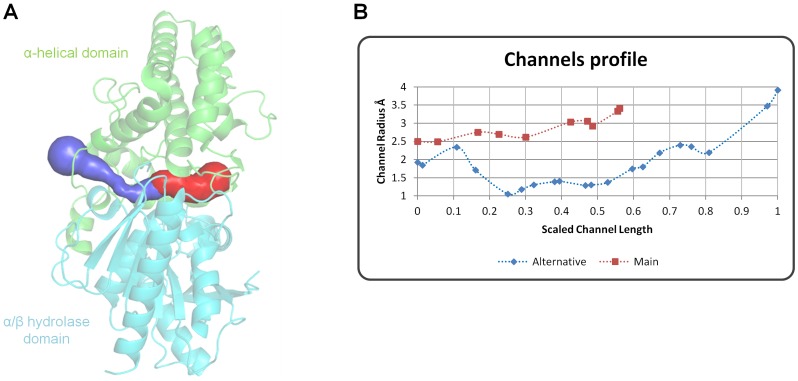
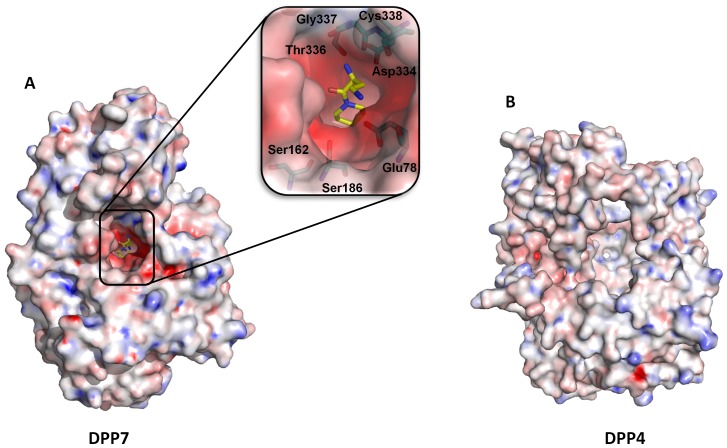
Proline-specific dipeptidyl peptidases (DPPs) are emerging targets for drug development. DPP4 inhibitors are approved in many countries, and other dipeptidyl peptidases are often referred to as DPP4 activity- and/or structure-homologues (DASH). Members of the DASH family have overlapping substrate specificities, and, even though they share low sequence identity, therapeutic or clinical cross-reactivity is a concern. Here, we report the structure of human DPP7 and its complex with a selective inhibitor Dab-Pip (L-2,4-diaminobutyryl-piperidinamide) and compare it with that of DPP4. Both enzymes share a common catalytic domain (α/β-hydrolase). The catalytic pocket is located in the interior of DPP7, deep inside the cleft between the two domains. Substrates might access the active site via a narrow tunnel. The DPP7 catalytic triad is completely conserved and comprises Ser162, Asp418 and His443 (corresponding to Ser630, Asp708 and His740 in DPP4), while other residues lining the catalytic pockets differ considerably. The "specificity domains" are structurally also completely different exhibiting a β-propeller fold in DPP4 compared to a rare, completely helical fold in DPP7. Comparing the structures of DPP7 and DPP4 allows the design of specific inhibitors and thus the development of less cross-reactive drugs. Furthermore, the reported DPP7 structures shed some light onto the evolutionary relationship of prolyl-specific peptidases through the analysis of the architectural organization of their domains.
Conflict of interest statement
Figures

References
-
- Yaron A, Naider F (1993) Proline-dependent structural and biological properties of peptides and proteins. Crit Rev Biochem Mol Biol 28: 31–81. - PubMed
-
- Rosenblum JS, Kozarich JW (2003) Prolyl peptidases: a serine protease subfamily with high potential for drug discovery. Curr Opin Chem Biol 7: 496–504. - PubMed
-
- Yazbeck R, Howarth GS, Abbott CA (2009) Dipeptidyl peptidase inhibitors, an emerging drug class for inflammatory disease? Trends Pharmacol Sci 30: 600–607. - PubMed
-
- Busek P, Malik R, Sedo A (2004) Dipeptidyl peptidase IV activity and/or structure homologues (DASH) and their substrates in cancer. Int J Biochem Cell Biol 36: 408–421. - PubMed
-
- Gorrell MD, Gysbers V, McCaughan GW (2001) CD26: a multifunctional integral membrane and secreted protein of activated lymphocytes. Scand J Immunol 54: 249–264. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
