

Landscape of transcription in human cells
- PMID: 22955620
- PMCID: PMC3684276
- DOI: 10.1038/nature11233
Landscape of transcription in human cells
Abstract
Eukaryotic cells make many types of primary and processed RNAs that are found either in specific subcellular compartments or throughout the cells. A complete catalogue of these RNAs is not yet available and their characteristic subcellular localizations are also poorly understood. Because RNA represents the direct output of the genetic information encoded by genomes and a significant proportion of a cell's regulatory capabilities are focused on its synthesis, processing, transport, modification and translation, the generation of such a catalogue is crucial for understanding genome function. Here we report evidence that three-quarters of the human genome is capable of being transcribed, as well as observations about the range and levels of expression, localization, processing fates, regulatory regions and modifications of almost all currently annotated and thousands of previously unannotated RNAs. These observations, taken together, prompt a redefinition of the concept of a gene.
Figures
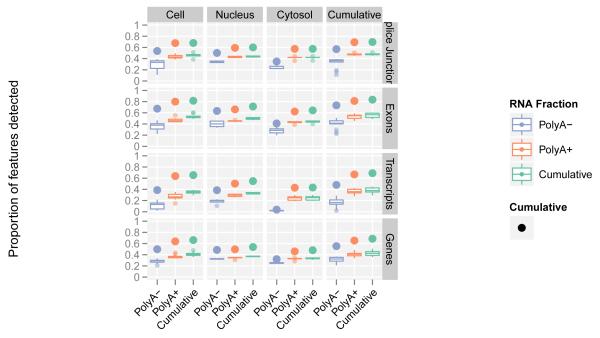
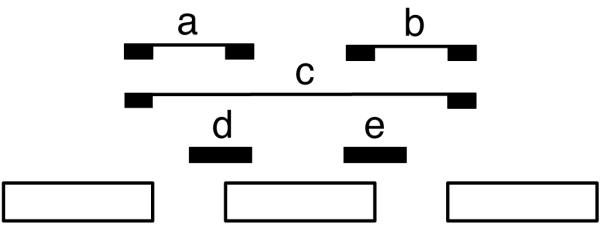
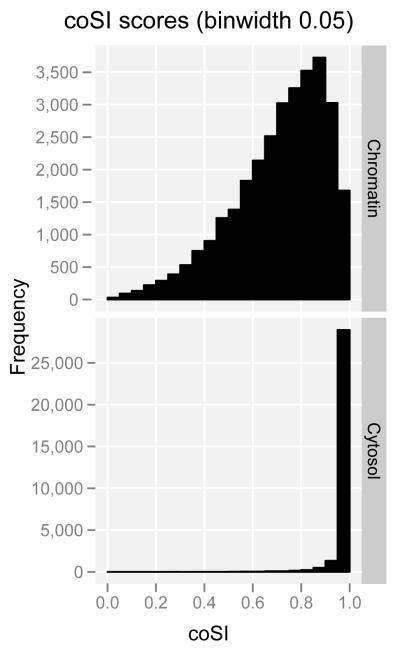
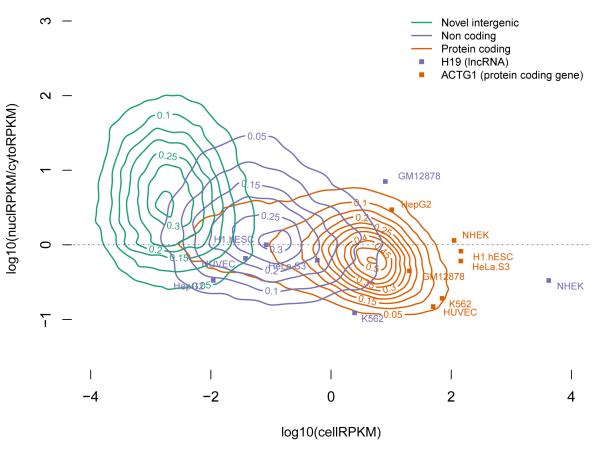
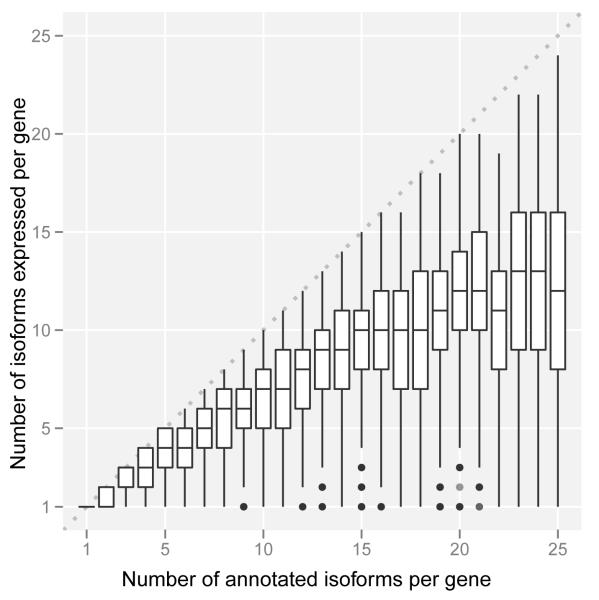
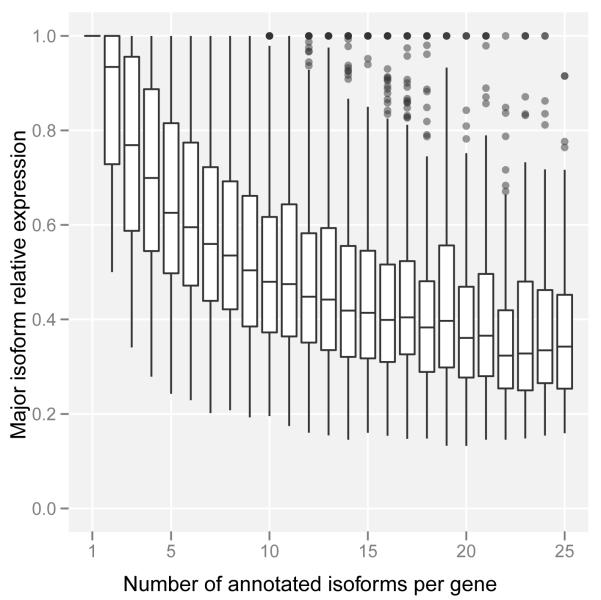
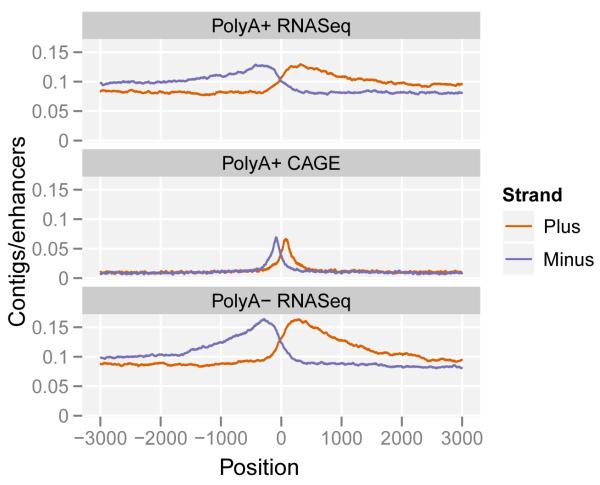
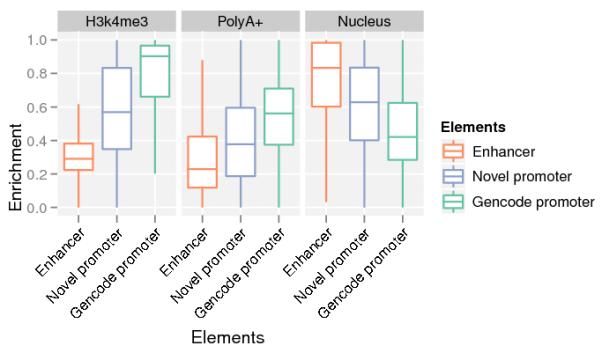
Comment in
-
Genomics: users' guide to the human genome.Nat Rev Genet. 2012 Oct;13(10):678. doi: 10.1038/nrg3329. Epub 2012 Sep 7. Nat Rev Genet. 2012. PMID: 22955793 No abstract available.
-
Discovering the complexity of the metazoan transcriptome.Genome Biol. 2014 Apr 29;15(4):112. doi: 10.1186/gb4172. Genome Biol. 2014. PMID: 25001157 Free PMC article.
References
-
- Mattick JS. Long noncoding RNAs in cell and developmental biology. Semin Cell Dev Biol. 2011;22:327. doi:S1084-9521(11)00077-2 [pii] 10.1016/j.semcdb.2011.05.002. - PubMed
-
- The ENCODE (ENCyclopedia Of DNA Elements) Project Science. 2004;306:636–640. doi:306/5696/636 [pii] 10.1126/science.1105136. - PubMed
-
- Kapranov P, et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316:1484–1488. doi:1138341 [pii] 10.1126/science.1138341. - PubMed
-
- Kapranov P, Willingham AT, Gingeras TR. Genome-wide transcription and the implications for genomic organization. Nat Rev Genet. 2007;8:413–423. doi:nrg2083 [pii] 10.1038/nrg2083. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
- RC2 HG005591/HG/NHGRI NIH HHS/United States
- U54 HG007004/HG/NHGRI NIH HHS/United States
- U54 HG004558/HG/NHGRI NIH HHS/United States
- U54HG004557/HG/NHGRI NIH HHS/United States
- U54 HG004557/HG/NHGRI NIH HHS/United States
- 249968/ERC_/European Research Council/International
- R01HG003700/HG/NHGRI NIH HHS/United States
- P30 CA045508/CA/NCI NIH HHS/United States
- U54 HG004576/HG/NHGRI NIH HHS/United States
- 062023/WT_/Wellcome Trust/United Kingdom
- U54HG004576/HG/NHGRI NIH HHS/United States
- U54 HG004555/HG/NHGRI NIH HHS/United States
- U54HG004558/HG/NHGRI NIH HHS/United States
- U01 HG003147/HG/NHGRI NIH HHS/United States
- 1RC2HG005591/HG/NHGRI NIH HHS/United States
- R37 GM062534/GM/NIGMS NIH HHS/United States
- R01 HG003700/HG/NHGRI NIH HHS/United States
- U54HG004555/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
