

RNA editing in the human ENCODE RNA-seq data
- PMID: 22955975
- PMCID: PMC3431480
- DOI: 10.1101/gr.134957.111
RNA editing in the human ENCODE RNA-seq data
Abstract
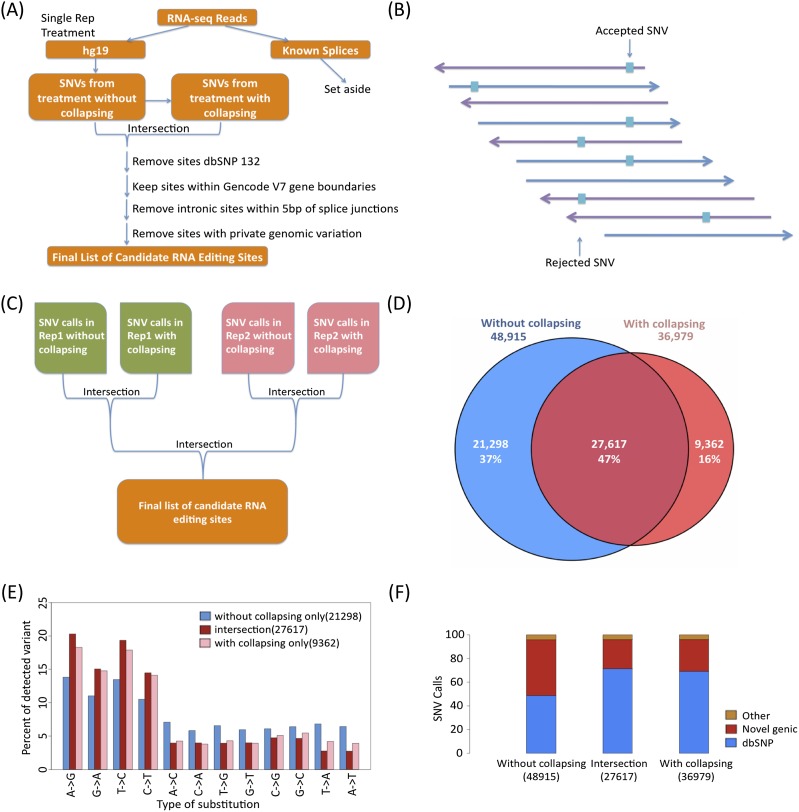
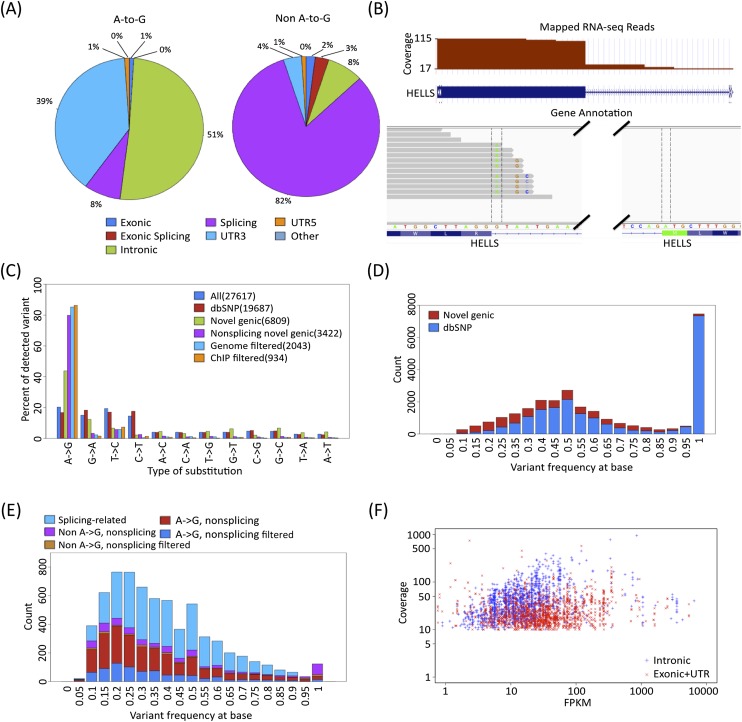
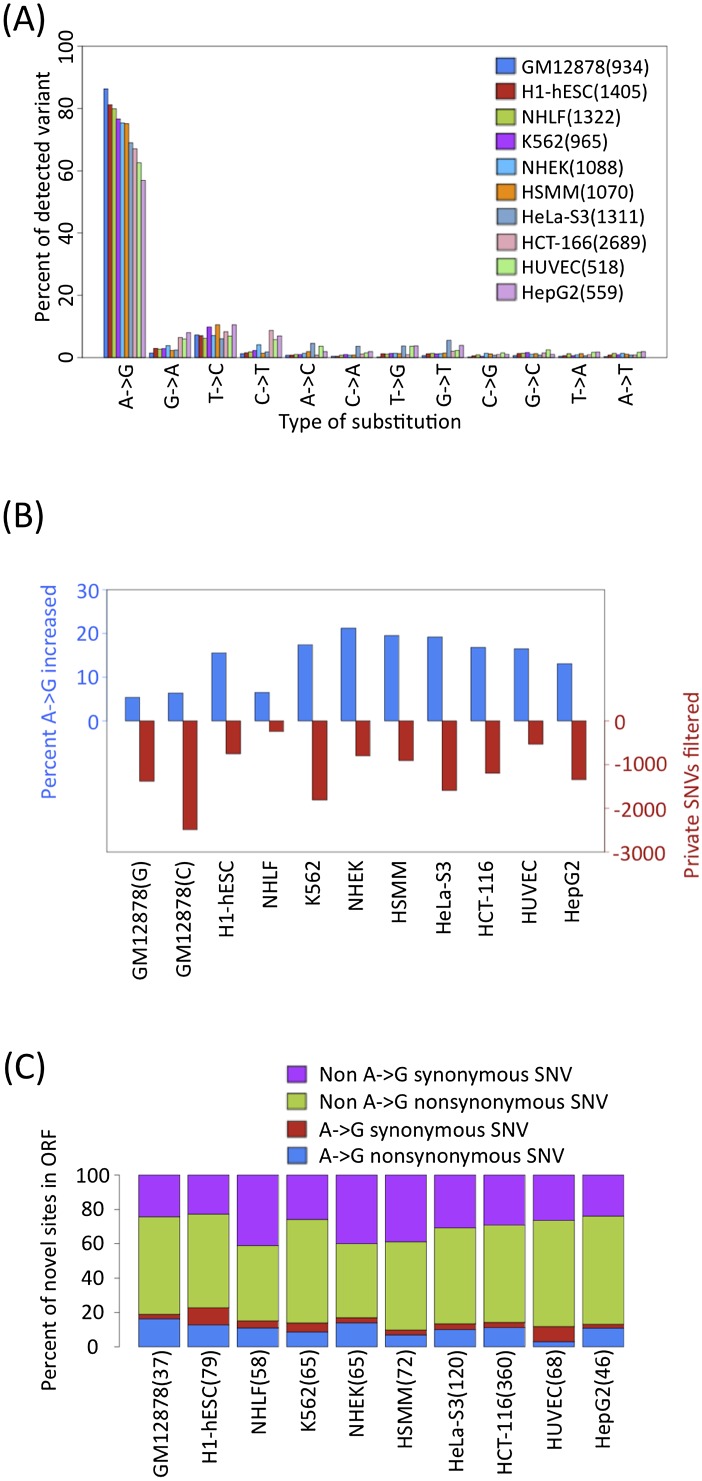
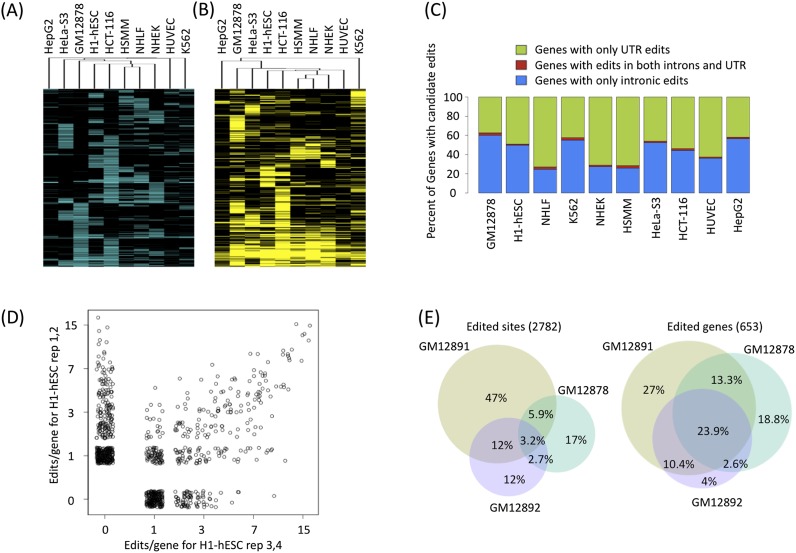
RNA-seq data can be mined for sequence differences relative to the reference genome to identify both genomic SNPs and RNA editing events. We analyzed the long, polyA-selected, unstranded, deeply sequenced RNA-seq data from the ENCODE Project across 14 human cell lines for candidate RNA editing events. On average, 43% of the RNA sequencing variants that are not in dbSNP and are within gene boundaries are A-to-G(I) RNA editing candidates. The vast majority of A-to-G(I) edits are located in introns and 3' UTRs, with only 123 located in protein-coding sequence. In contrast, the majority of non-A-to-G variants (60%-80%) map near exon boundaries and have the characteristics of splice-mapping artifacts. After filtering out all candidates with evidence of private genomic variation using genome resequencing or ChIP-seq data, we find that up to 85% of the high-confidence RNA variants are A-to-G(I) editing candidates. Genes with A-to-G(I) edits are enriched in Gene Ontology terms involving cell division, viral defense, and translation. The distribution and character of the remaining non-A-to-G variants closely resemble known SNPs. We find no reproducible A-to-G(I) edits that result in nonsynonymous substitutions in all three lymphoblastoid cell lines in our study, unlike RNA editing in the brain. Given that only a fraction of sites are reproducibly edited in multiple cell lines and that we find a stronger association of editing and specific genes suggests that the editing of the transcript is more important than the editing of any individual site.
Figures
References
-
- Burns CM, Chu H, Rueter SM, Hutchinson LK, Canton H, Sanders-Bush E, Emeson RB 1997. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature 387: 303–308 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical