

Understanding transcriptional regulation by integrative analysis of transcription factor binding data
- PMID: 22955978
- PMCID: PMC3431483
- DOI: 10.1101/gr.136838.111
Understanding transcriptional regulation by integrative analysis of transcription factor binding data
Abstract
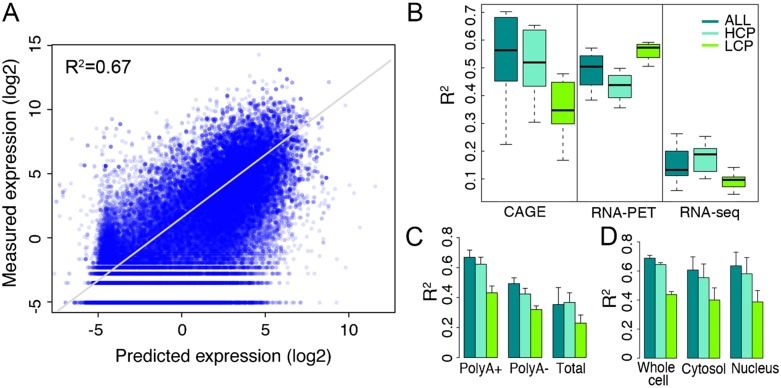
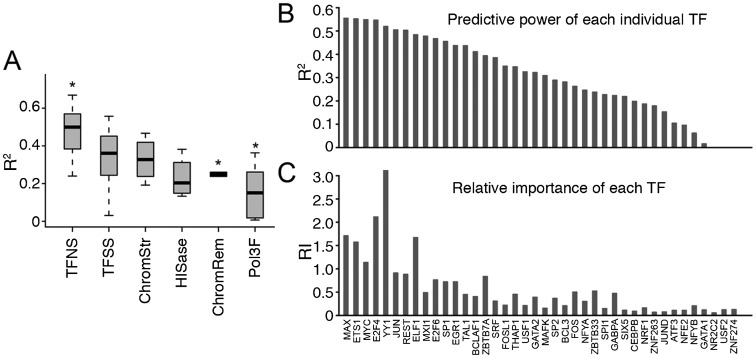
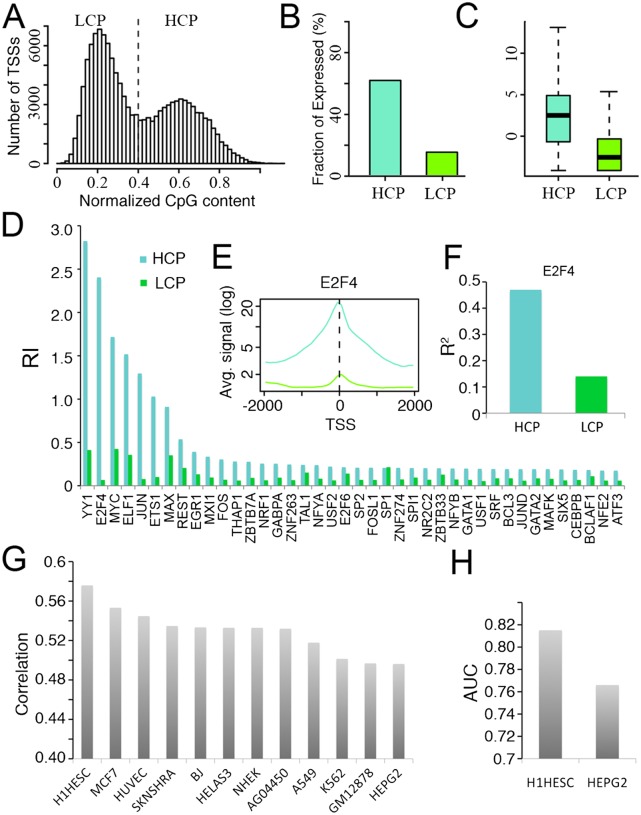
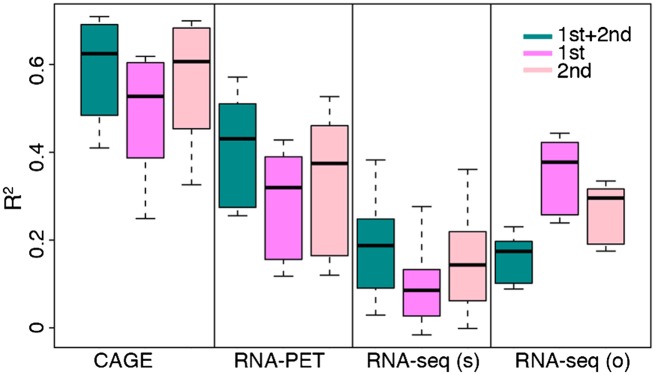
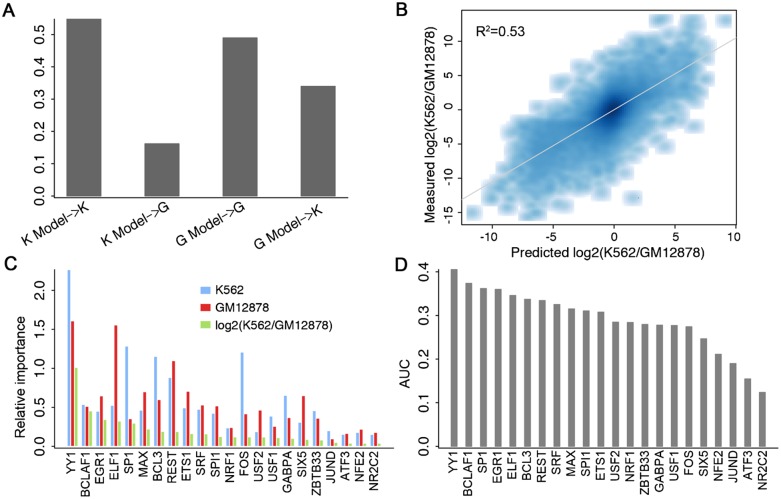
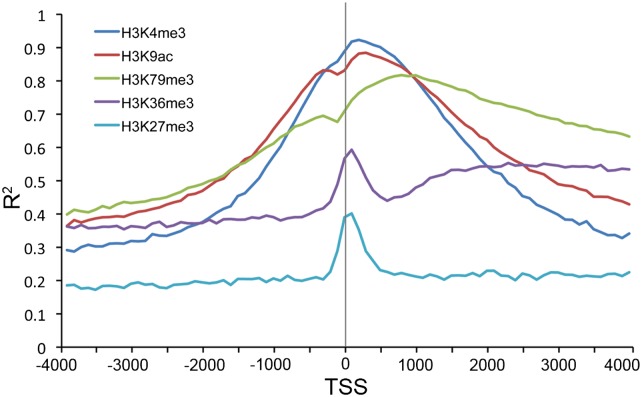
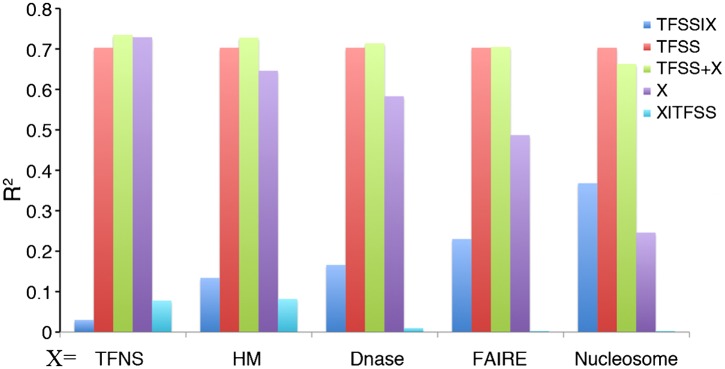
Statistical models have been used to quantify the relationship between gene expression and transcription factor (TF) binding signals. Here we apply the models to the large-scale data generated by the ENCODE project to study transcriptional regulation by TFs. Our results reveal a notable difference in the prediction accuracy of expression levels of transcription start sites (TSSs) captured by different technologies and RNA extraction protocols. In general, the expression levels of TSSs with high CpG content are more predictable than those with low CpG content. For genes with alternative TSSs, the expression levels of downstream TSSs are more predictable than those of the upstream ones. Different TF categories and specific TFs vary substantially in their contributions to predicting expression. Between two cell lines, the differential expression of TSS can be precisely reflected by the difference of TF-binding signals in a quantitative manner, arguing against the conventional on-and-off model of TF binding. Finally, we explore the relationships between TF-binding signals and other chromatin features such as histone modifications and DNase hypersensitivity for determining expression. The models imply that these features regulate transcription in a highly coordinated manner.
Figures

References
-
- Babu MM, Luscombe NM, Aravind L, Gerstein M, Teichmann SA 2004. Structure and evolution of transcriptional regulatory networks. Curr Opin Struct Biol 14: 283–291 - PubMed
-
- Biggin MD 2011. Animal transcription networks as highly connected, quantitative continua. Dev Cell 21: 611–626 - PubMed
-
- Breiman L 2001. Random Forests. Mach Learn 45: 5–32
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous