

A highly integrated and complex PPARGC1A transcription factor binding network in HepG2 cells
- PMID: 22955979
- PMCID: PMC3431484
- DOI: 10.1101/gr.127761.111
A highly integrated and complex PPARGC1A transcription factor binding network in HepG2 cells
Abstract
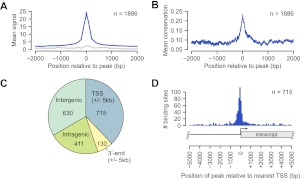
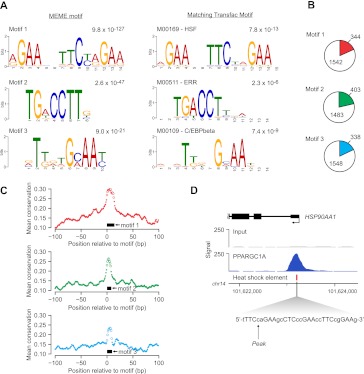
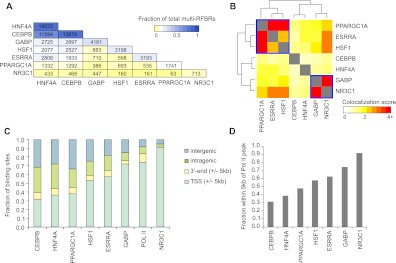
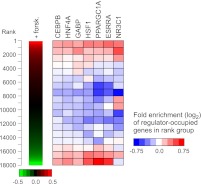
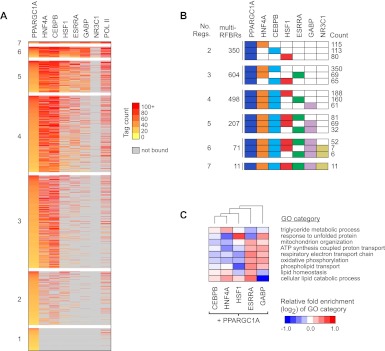
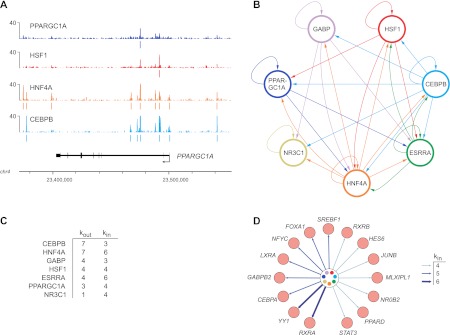
PPARGC1A is a transcriptional coactivator that binds to and coactivates a variety of transcription factors (TFs) to regulate the expression of target genes. PPARGC1A plays a pivotal role in regulating energy metabolism and has been implicated in several human diseases, most notably type II diabetes. Previous studies have focused on the interplay between PPARGC1A and individual TFs, but little is known about how PPARGC1A combines with all of its partners across the genome to regulate transcriptional dynamics. In this study, we describe a core PPARGC1A transcriptional regulatory network operating in HepG2 cells treated with forskolin. We first mapped the genome-wide binding sites of PPARGC1A using chromatin-IP followed by high-throughput sequencing (ChIP-seq) and uncovered overrepresented DNA sequence motifs corresponding to known and novel PPARGC1A network partners. We then profiled six of these site-specific TF partners using ChIP-seq and examined their network connectivity and combinatorial binding patterns with PPARGC1A. Our analysis revealed extensive overlap of targets including a novel link between PPARGC1A and HSF1, a TF regulating the conserved heat shock response pathway that is misregulated in diabetes. Importantly, we found that different combinations of TFs bound to distinct functional sets of genes, thereby helping to reveal the combinatorial regulatory code for metabolic and other cellular processes. In addition, the different TFs often bound near the promoters and coding regions of each other's genes suggesting an intricate network of interdependent regulation. Overall, our study provides an important framework for understanding the systems-level control of metabolic gene expression in humans.
Figures
References
-
- Bailey TL, Elkan C 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2: 28–36 - PubMed
-
- Barabasi AL, Oltvai ZN 2004. Network biology: Understanding the cell's functional organization. Nat Rev Genet 5: 101–113 - PubMed
-
- Blais A, Dynlacht BD 2005. Constructing transcriptional regulatory networks. Genes Dev 19: 1499–1511 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous