

Personal and population genomics of human regulatory variation
- PMID: 22955981
- PMCID: PMC3431486
- DOI: 10.1101/gr.134890.111
Personal and population genomics of human regulatory variation
Abstract
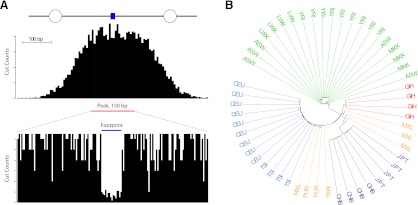
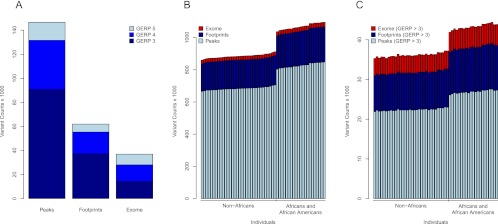
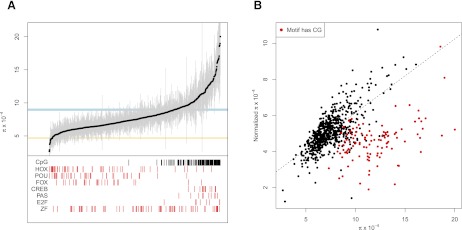
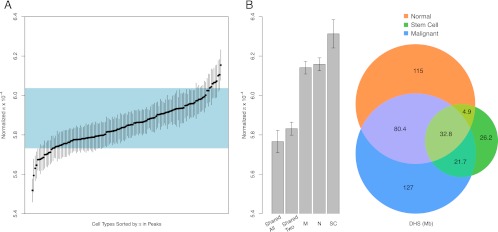
The characteristics and evolutionary forces acting on regulatory variation in humans remains elusive because of the difficulty in defining functionally important noncoding DNA. Here, we combine genome-scale maps of regulatory DNA marked by DNase I hypersensitive sites (DHSs) from 138 cell and tissue types with whole-genome sequences of 53 geographically diverse individuals in order to better delimit the patterns of regulatory variation in humans. We estimate that individuals likely harbor many more functionally important variants in regulatory DNA compared with protein-coding regions, although they are likely to have, on average, smaller effect sizes. Moreover, we demonstrate that there is significant heterogeneity in the level of functional constraint in regulatory DNA among different cell types. We also find marked variability in functional constraint among transcription factor motifs in regulatory DNA, with sequence motifs for major developmental regulators, such as HOX proteins, exhibiting levels of constraint comparable to protein-coding regions. Finally, we perform a genome-wide scan of recent positive selection and identify hundreds of novel substrates of adaptive regulatory evolution that are enriched for biologically interesting pathways such as melanogenesis and adipocytokine signaling. These data and results provide new insights into patterns of regulatory variation in individuals and populations and demonstrate that a large proportion of functionally important variation lies beyond the exome.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources