

Predicting cell-type-specific gene expression from regions of open chromatin
- PMID: 22955983
- PMCID: PMC3431488
- DOI: 10.1101/gr.135129.111
Predicting cell-type-specific gene expression from regions of open chromatin
Abstract
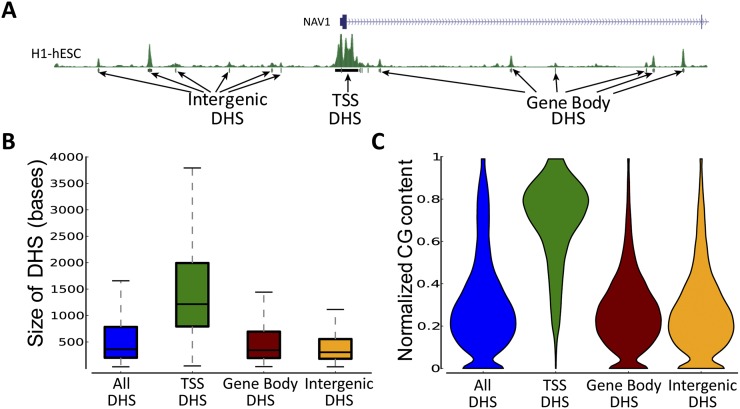
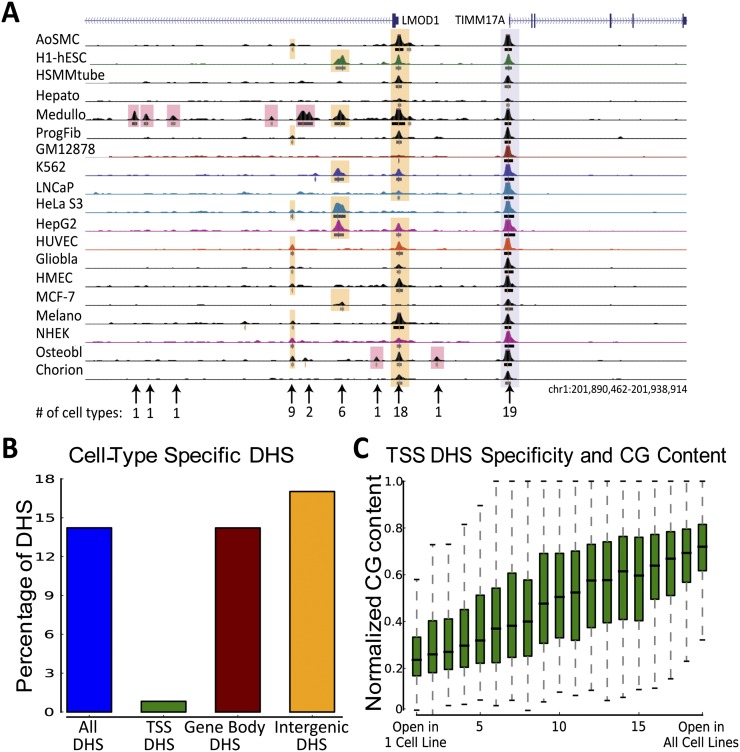
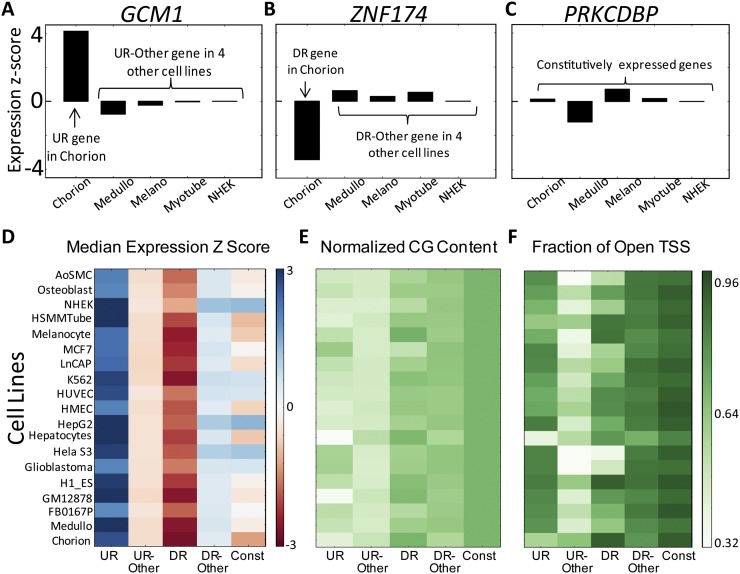
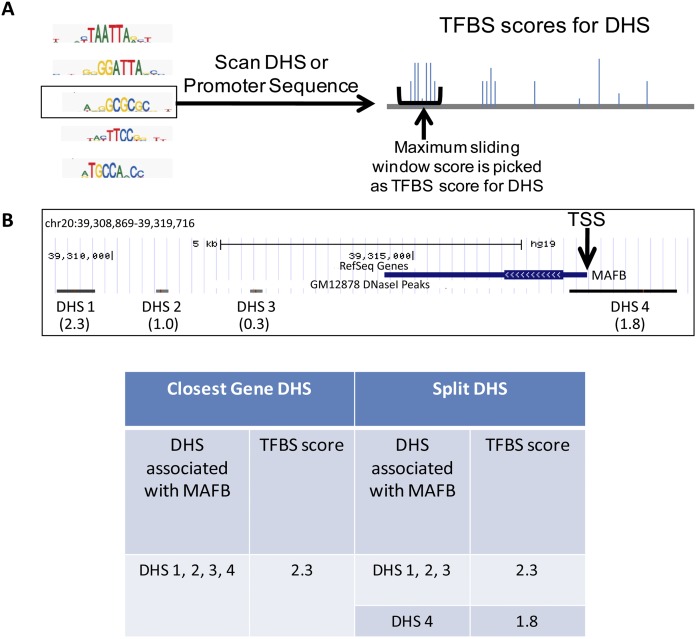
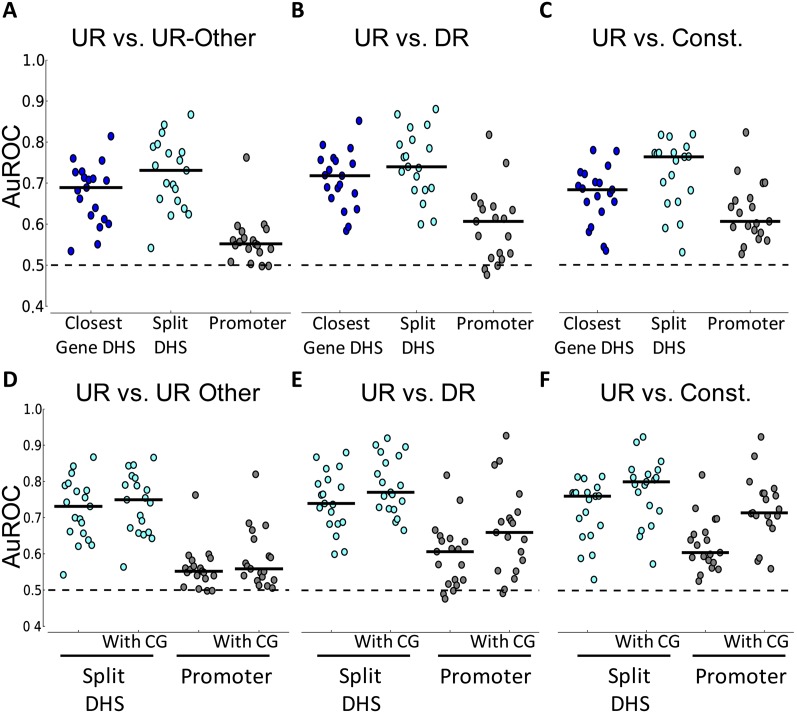
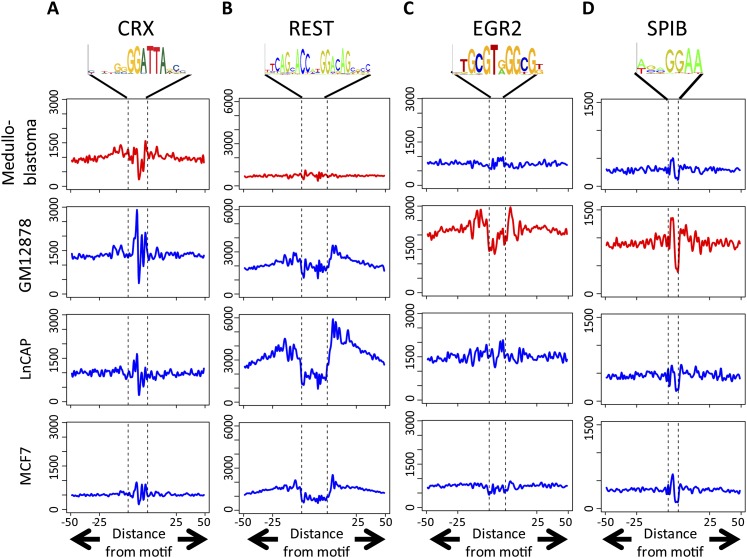
Complex patterns of cell-type-specific gene expression are thought to be achieved by combinatorial binding of transcription factors (TFs) to sequence elements in regulatory regions. Predicting cell-type-specific expression in mammals has been hindered by the oftentimes unknown location of distal regulatory regions. To alleviate this bottleneck, we used DNase-seq data from 19 diverse human cell types to identify proximal and distal regulatory elements at genome-wide scale. Matched expression data allowed us to separate genes into classes of cell-type-specific up-regulated, down-regulated, and constitutively expressed genes. CG dinucleotide content and DNA accessibility in the promoters of these three classes of genes displayed substantial differences, highlighting the importance of including these aspects in modeling gene expression. We associated DNase I hypersensitive sites (DHSs) with genes, and trained classifiers for different expression patterns. TF sequence motif matches in DHSs provided a strong performance improvement in predicting gene expression over the typical baseline approach of using proximal promoter sequences. In particular, we achieved competitive performance when discriminating up-regulated genes from different cell types or genes up- and down-regulated under the same conditions. We identified previously known and new candidate cell-type-specific regulators. The models generated testable predictions of activating or repressive functions of regulators. DNase I footprints for these regulators were indicative of their direct binding to DNA. In summary, we successfully used information of open chromatin obtained by a single assay, DNase-seq, to address the problem of predicting cell-type-specific gene expression in mammalian organisms directly from regulatory sequence.
Figures
References
-
- Aerts S, Van Loo P, Thijs G, Moreau Y, De Moor B 2003. Computational detection of cis-regulatory modules. Bioinformatics (Suppl 2) 19: ii5–ii14 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous