

Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors
- PMID: 22955990
- PMCID: PMC3431495
- DOI: 10.1101/gr.139105.112
Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors
Abstract
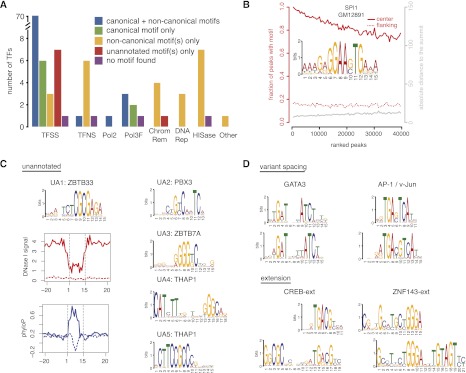
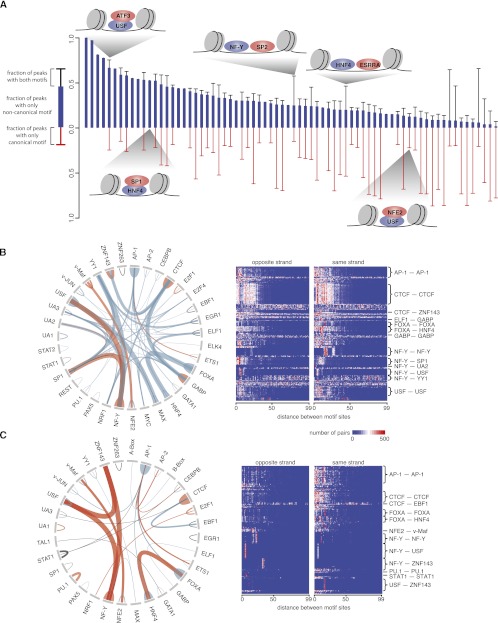
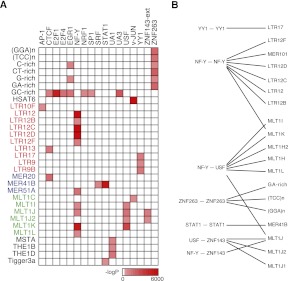
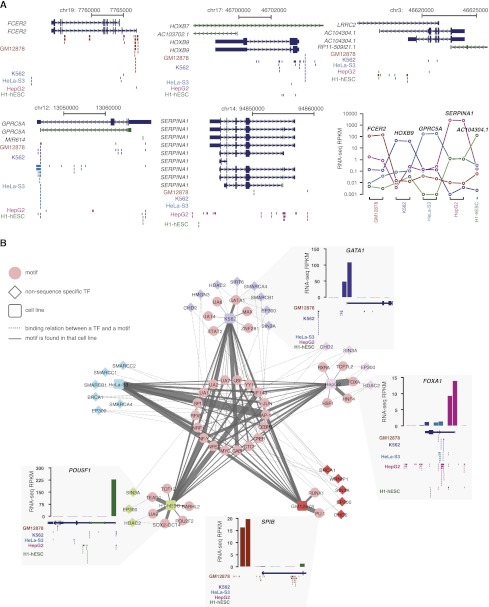
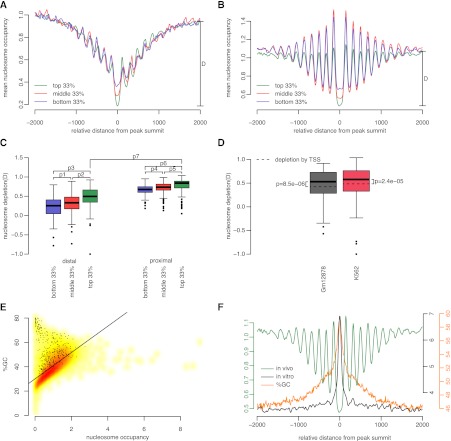
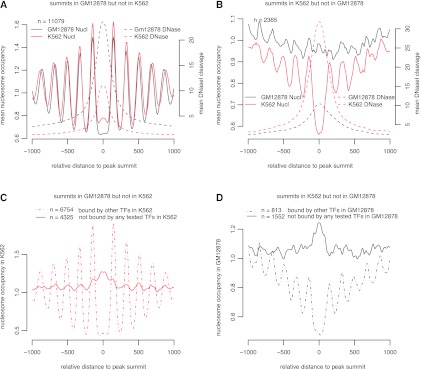
Chromatin immunoprecipitation coupled with high-throughput sequencing (ChIP-seq) has become the dominant technique for mapping transcription factor (TF) binding regions genome-wide. We performed an integrative analysis centered around 457 ChIP-seq data sets on 119 human TFs generated by the ENCODE Consortium. We identified highly enriched sequence motifs in most data sets, revealing new motifs and validating known ones. The motif sites (TF binding sites) are highly conserved evolutionarily and show distinct footprints upon DNase I digestion. We frequently detected secondary motifs in addition to the canonical motifs of the TFs, indicating tethered binding and cobinding between multiple TFs. We observed significant position and orientation preferences between many cobinding TFs. Genes specifically expressed in a cell line are often associated with a greater occurrence of nearby TF binding in that cell line. We observed cell-line-specific secondary motifs that mediate the binding of the histone deacetylase HDAC2 and the enhancer-binding protein EP300. TF binding sites are located in GC-rich, nucleosome-depleted, and DNase I sensitive regions, flanked by well-positioned nucleosomes, and many of these features show cell type specificity. The GC-richness may be beneficial for regulating TF binding because, when unoccupied by a TF, these regions are occupied by nucleosomes in vivo. We present the results of our analysis in a TF-centric web repository Factorbook (http://factorbook.org) and will continually update this repository as more ENCODE data are generated.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous