

Selective activation of ATF6 and PERK endoplasmic reticulum stress signaling pathways prevent mutant rhodopsin accumulation
- PMID: 22956602
- PMCID: PMC3474590
- DOI: 10.1167/iovs.12-10222
Selective activation of ATF6 and PERK endoplasmic reticulum stress signaling pathways prevent mutant rhodopsin accumulation
Abstract
Purpose: Many rhodopsin mutations that cause retinitis pigmentosa produce misfolded rhodopsin proteins that are retained within the endoplasmic reticulum (ER) and cause photoreceptor cell death. Activating transcription factor 6 (ATF6) and protein kinase RNA-like endoplasmic reticulum kinase (PERK) control intracellular signaling pathways that maintain ER homeostasis. The aim of this study was to investigate how ATF6 and PERK signaling affected misfolded rhodopsin in cells, which could identify new molecular therapies to treat retinal diseases associated with ER protein misfolding.
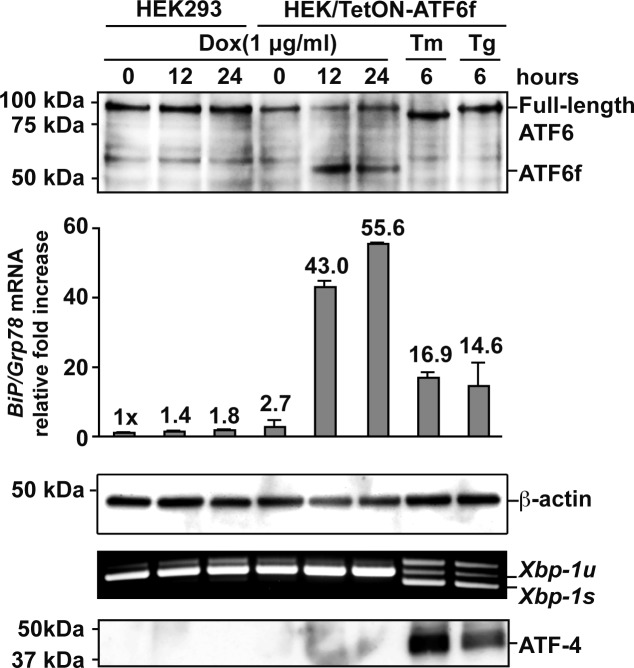
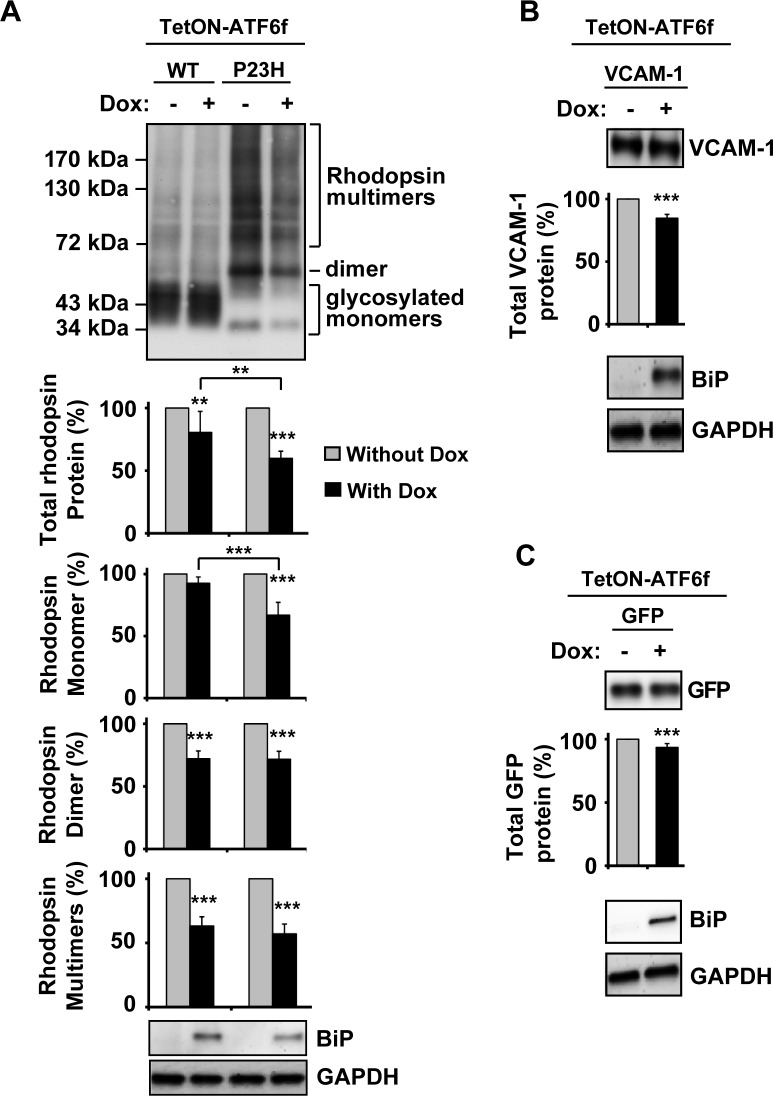
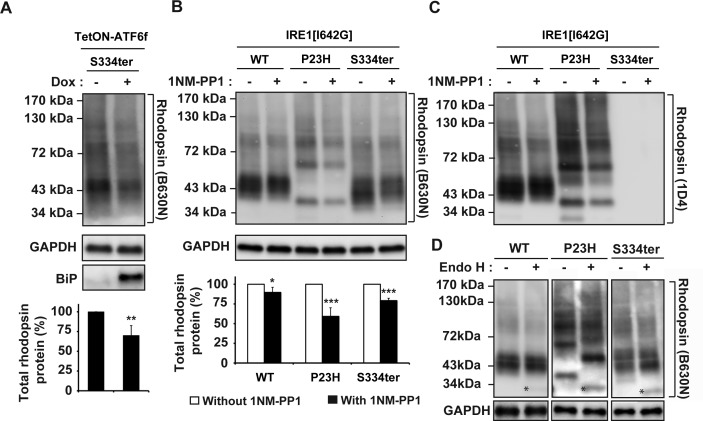
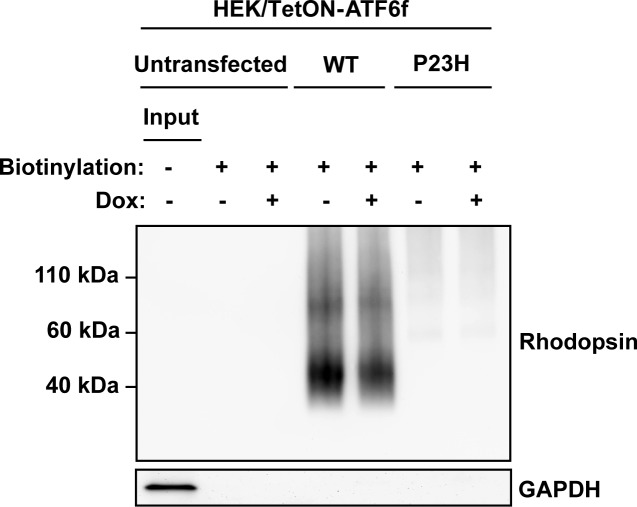
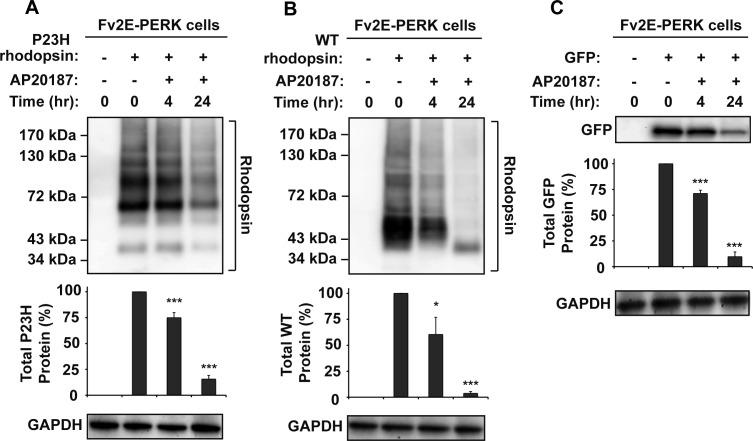
Methods: To examine the effect of ATF6 on rhodopsin, wild-type (WT) or mutant rhodopsins were expressed in cells expressing inducible human ATF6f, the transcriptional activator domain of ATF6. Induction of ATF6f synthesis rapidly activated downstream genes. To examine PERK's effect on rhodopsin, WT or mutant rhodopsins were expressed in cells expressing a genetically altered PERK protein, Fv2E-PERK. Addition of the dimerizing molecule (AP20187) rapidly activated Fv2E-PERK and downstream genes. By use of these strategies, it was examined how selective ATF6 or PERK signaling affected the fate of WT and mutant rhodopsins.
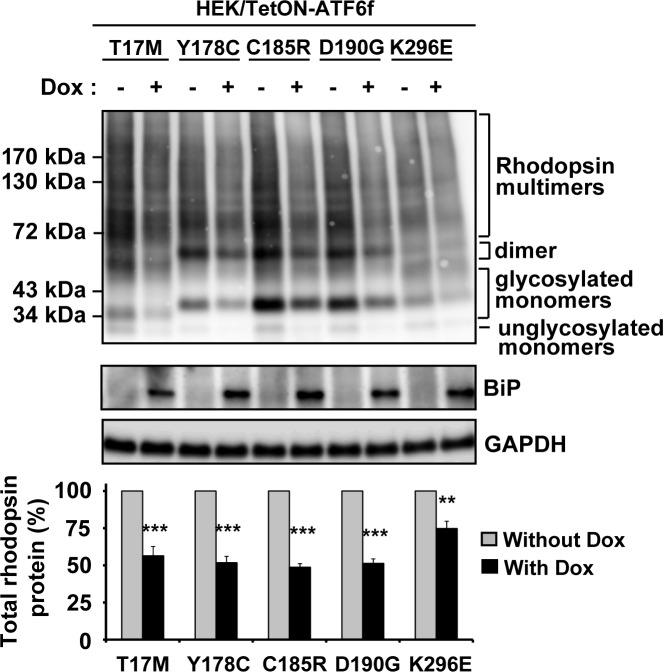
Results: ATF6 significantly reduced T17M, P23H, Y178C, C185R, D190G, K296E, and S334ter rhodopsin protein levels in the cells with minimal effects on monomeric WT rhodopsin protein levels. By contrast, the PERK pathway reduced both levels of WT, mutant rhodopsins, and many other proteins in the cell.
Conclusions: This study indicates that selectively activating ATF6 or PERK prevents mutant rhodopsin from accumulating in cells. ATF6 signaling may be especially useful in treating retinal degenerative diseases arising from rhodopsin misfolding by preferentially clearing mutant rhodopsin and abnormal rhodopsin aggregates.
Conflict of interest statement
Disclosure:
Figures
Comment in
-
Unpicking the UPR.Invest Ophthalmol Vis Sci. 2012 Oct 1;53(11):7167. doi: 10.1167/iovs.12-11004. Invest Ophthalmol Vis Sci. 2012. PMID: 23065632 No abstract available.
References
-
- Krauss HR, Heckenlively JR. Visual field changes in cone-rod degenerations. Arch Ophthalmol. 1982;100:1784–1790 - PubMed
-
- Berson EL. Retinitis pigmentosa. The Friedenwald Lecture. Invest Ophthalmol Vis Sci. 1993;34:1659–1676 - PubMed
-
- Kaushal S, Khorana HG. Structure and function in rhodopsin. 7. Point mutations associated with autosomal dominant retinitis pigmentosa. Biochemistry. 1994;33:6121–6128 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
