

A zebrafish model of PMM2-CDG reveals altered neurogenesis and a substrate-accumulation mechanism for N-linked glycosylation deficiency
- PMID: 22956764
- PMCID: PMC3484097
- DOI: 10.1091/mbc.E12-05-0411
A zebrafish model of PMM2-CDG reveals altered neurogenesis and a substrate-accumulation mechanism for N-linked glycosylation deficiency
Abstract
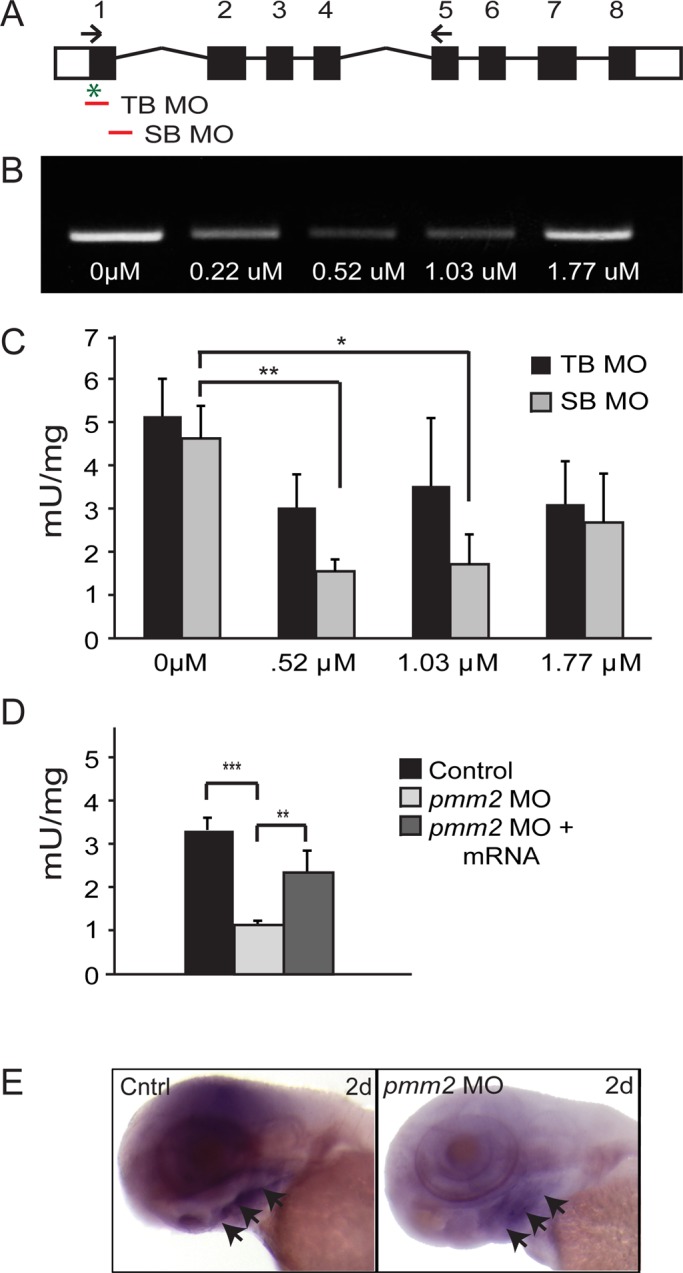
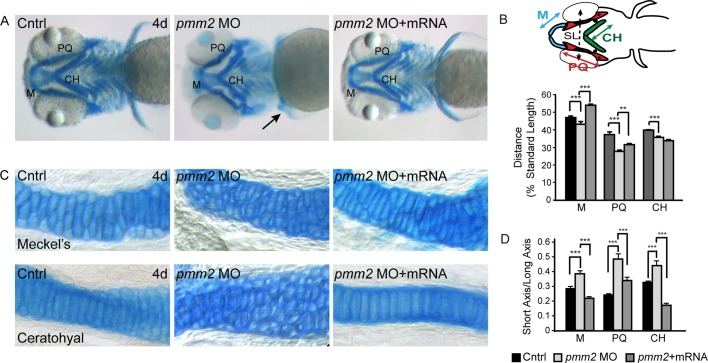
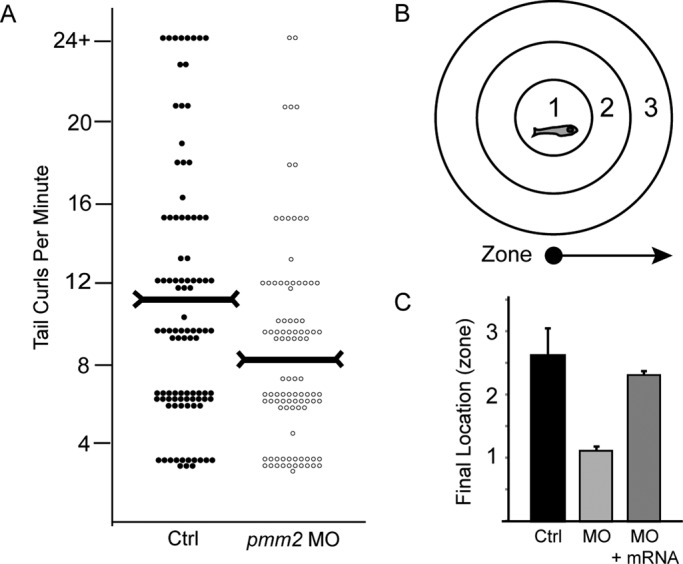
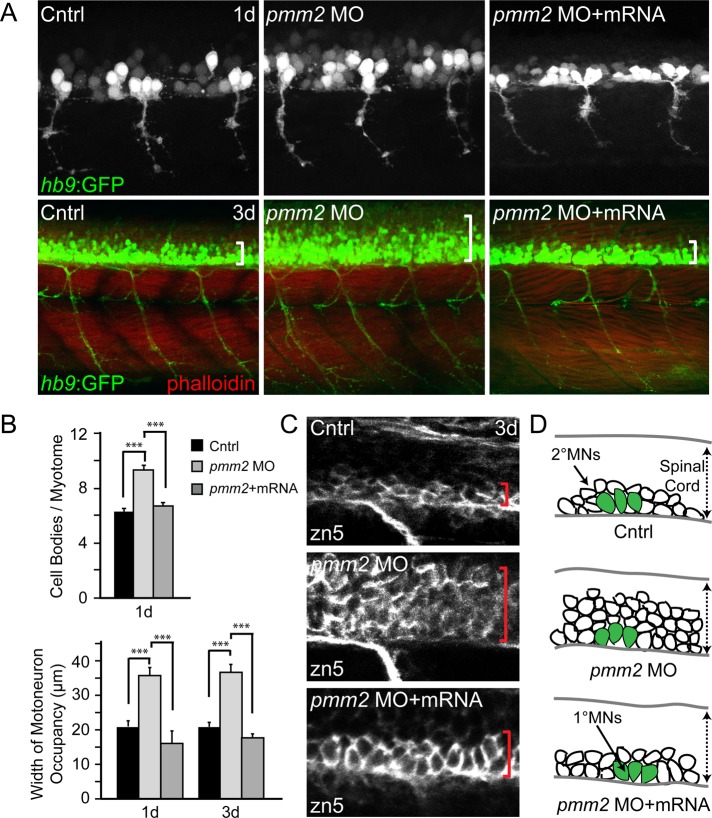
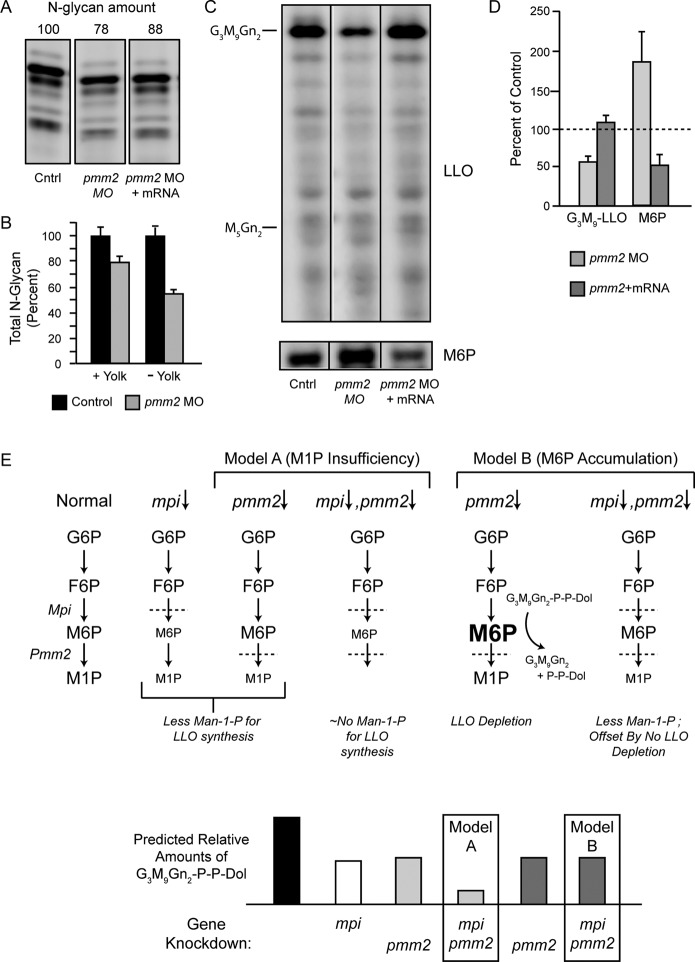
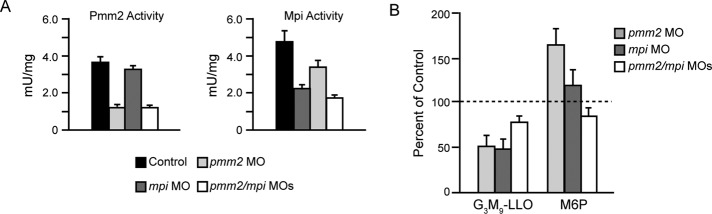
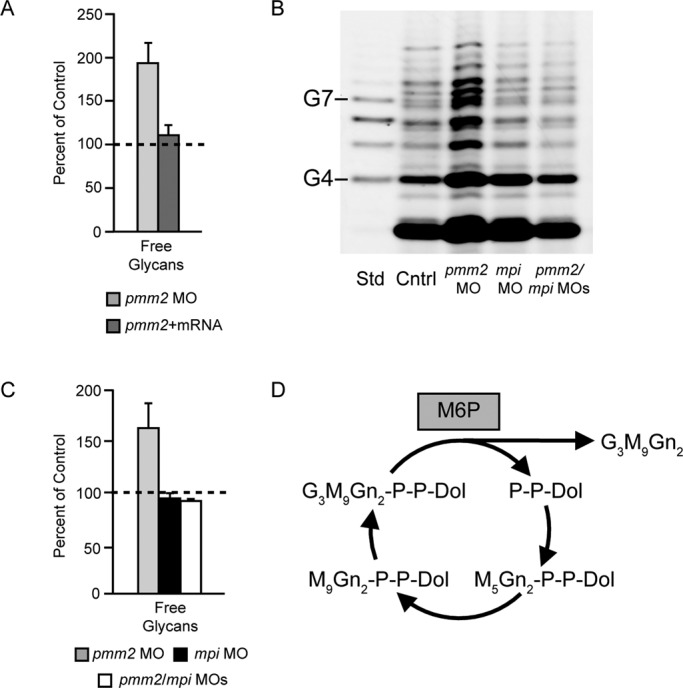
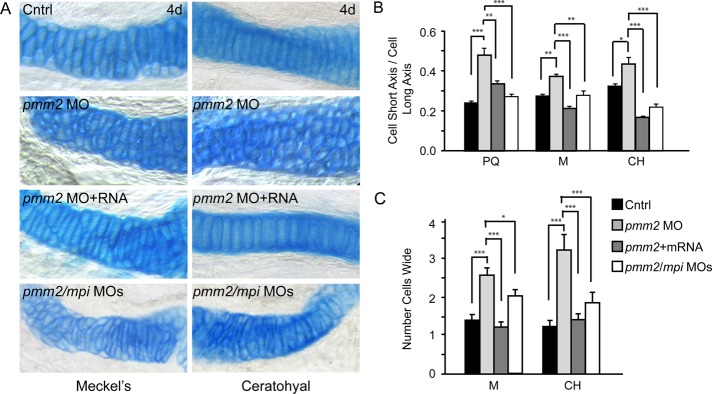
Congenital disorder of glycosylation (PMM2-CDG) results from mutations in pmm2, which encodes the phosphomannomutase (Pmm) that converts mannose-6-phosphate (M6P) to mannose-1-phosphate (M1P). Patients have wide-spectrum clinical abnormalities associated with impaired protein N-glycosylation. Although it has been widely proposed that Pmm2 deficiency depletes M1P, a precursor of GDP-mannose, and consequently suppresses lipid-linked oligosaccharide (LLO) levels needed for N-glycosylation, these deficiencies have not been demonstrated in patients or any animal model. Here we report a morpholino-based PMM2-CDG model in zebrafish. Morphant embryos had developmental abnormalities consistent with PMM2-CDG patients, including craniofacial defects and impaired motility associated with altered motor neurogenesis within the spinal cord. Significantly, global N-linked glycosylation and LLO levels were reduced in pmm2 morphants. Although M1P and GDP-mannose were below reliable detection/quantification limits, Pmm2 depletion unexpectedly caused accumulation of M6P, shown earlier to promote LLO cleavage in vitro. In pmm2 morphants, the free glycan by-products of LLO cleavage increased nearly twofold. Suppression of the M6P-synthesizing enzyme mannose phosphate isomerase within the pmm2 background normalized M6P levels and certain aspects of the craniofacial phenotype and abrogated pmm2-dependent LLO cleavage. In summary, we report the first zebrafish model of PMM2-CDG and uncover novel cellular insights not possible with other systems, including an M6P accumulation mechanism for underglycosylation.
Figures
References
-
- Chu J, Mir A, Gao N, Rosa S, Monson C, Sharma V, Steet R, Freeze HH, Lehrman MA, Sadler KC. A zebrafish model of congenital disorders of glycosylation with phosphomannose isomerase deficiency reveals an early opportunity for corrective mannose supplementation. Dis Model Mech. 2012 DOI: 10.1242/dmm.010116. - PMC - PubMed
-
- Coman D, Irving M, Kannu P, Jaeken J, Savarirayan R. The skeletal manifestations of the congenital disorders of glycosylation. Clin Genet. 2008;73:507–515. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
