

Rapamycin delays disease onset and prevents PrP plaque deposition in a mouse model of Gerstmann-Sträussler-Scheinker disease
- PMID: 22956830
- PMCID: PMC3752082
- DOI: 10.1523/JNEUROSCI.6189-11.2012
Rapamycin delays disease onset and prevents PrP plaque deposition in a mouse model of Gerstmann-Sträussler-Scheinker disease
Abstract
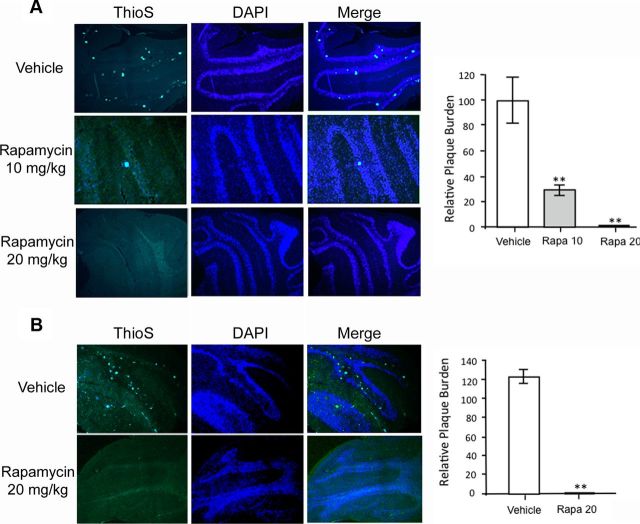
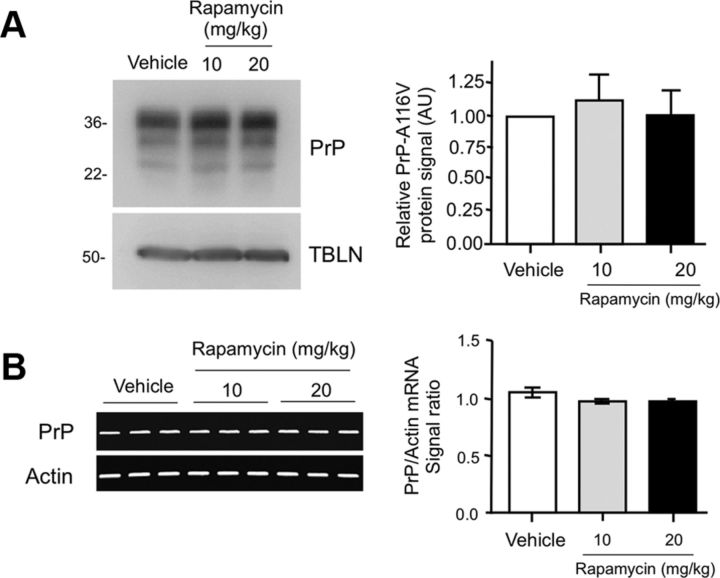
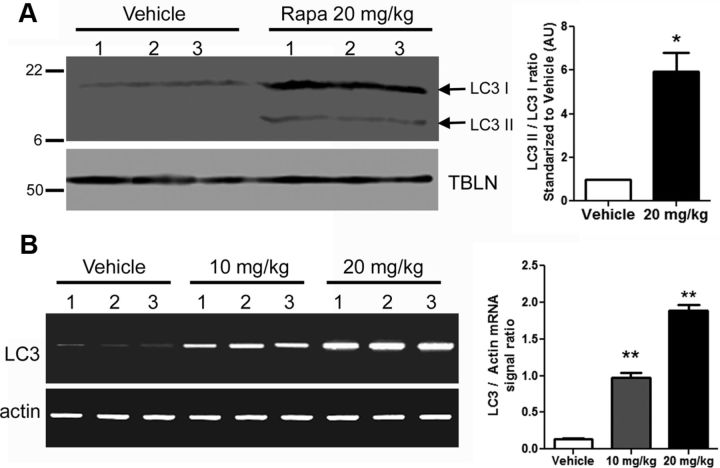
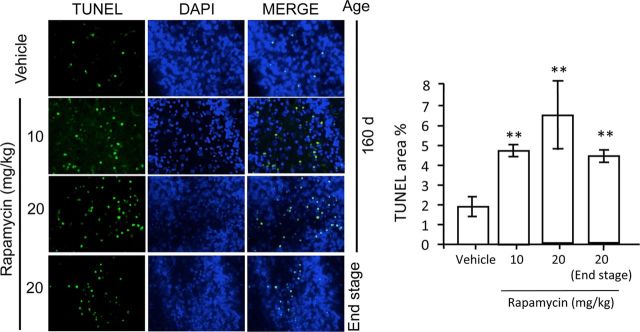
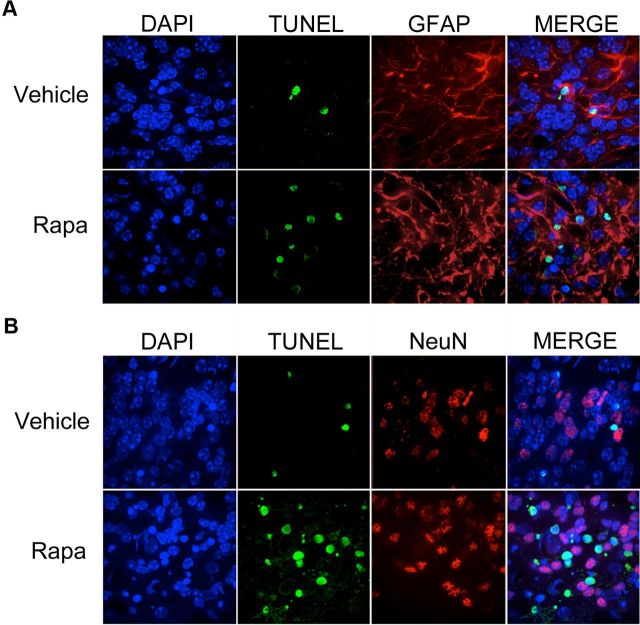
Autophagy is a cell survival response to nutrient deprivation that delivers cellular components to lysosomes for digestion. In recent years, autophagy has also been shown to assist in the degradation of misfolded proteins linked to neurodegenerative disease (Ross and Poirier, 2004). In support of this, rapamycin, an autophagy inducer, improves the phenotype of several animal models of neurodegenerative disease. Our Tg(PrP-A116V) mice model Gerstmann-Sträussler-Scheinker disease (GSS), a genetic prion disease characterized by prominent ataxia and extracellular PrP amyloid plaque deposits in brain (Yang et al., 2009). To determine whether autophagy induction can mitigate the development of GSS, Tg(PrP-A116V) mice were chronically treated with 10 or 20 mg/kg rapamycin intraperitoneally thrice weekly, beginning at 6 weeks of age. We observed a dose-related delay in disease onset, a reduction in symptom severity, and an extension of survival in rapamycin-treated Tg(PrP-A116V) mice. Coincident with this response was an increase in the autophagy-specific marker LC3II, a reduction in insoluble PrP-A116V, and a near-complete absence of PrP amyloid plaques in the brain. An increase in glial cell apoptosis of unclear significance was also detected. These findings suggest autophagy induction enhances elimination of misfolded PrP before its accumulation in plaques. Because ataxia persisted in these mice despite the absence of plaque deposits, our findings also suggest that PrP plaque pathology, a histopathological marker for the diagnosis of GSS, is not essential for the GSS phenotype.
Figures
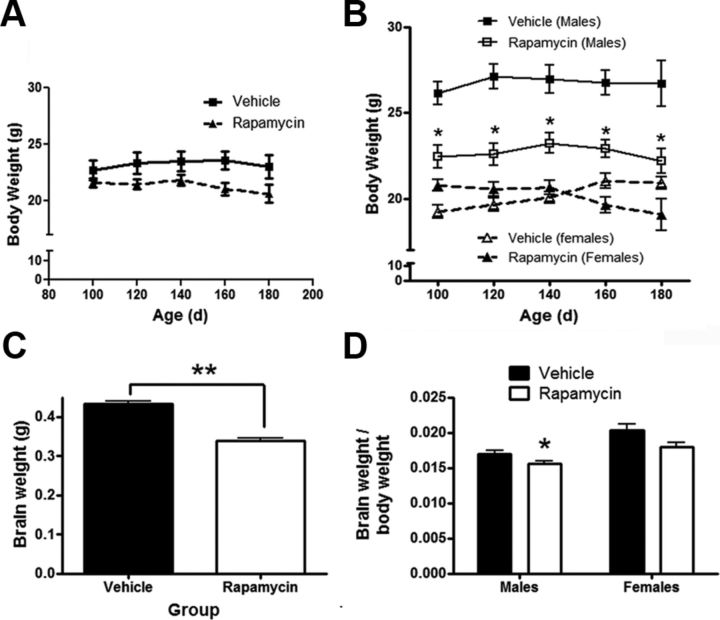
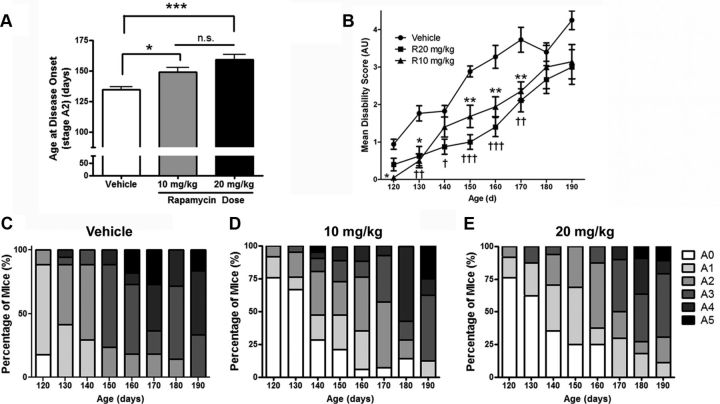
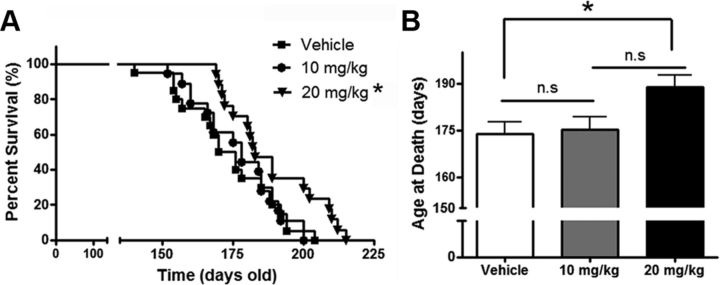
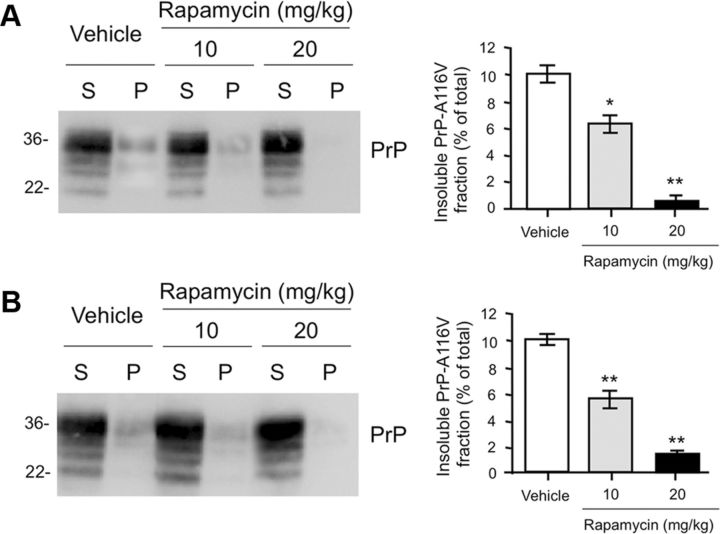
References
-
- Aguib Y, Heiseke A, Gilch S, Riemer C, Baier M, Schätzl HM, Ertmer A. Autophagy induction by trehalose counteracts cellular prion infection. Autophagy. 2009;5:361–369. - PubMed
-
- Aguzzi A, O'Connor T. Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nat Rev Drug Discov. 2010;9:237–248. - PubMed
-
- Aguzzi A, Heppner FL, Heikenwalder M, Prinz M, Mertz K, Seeger H, Glatzel M. Immune system and peripheral nerves in propagation of prions to CNS. Br Med Bull. 2003;66:141–159. - PubMed
-
- Berger Z, Ravikumar B, Menzies FM, Oroz LG, Underwood BR, Pangalos MN, Schmitt I, Wullner U, Evert BO, O'Kane CJ, Rubinsztein DC. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet. 2006;15:433–442. - PubMed
-
- Bounhar Y, Zhang Y, Goodyer CG, LeBlanc A. Prion protein protects human neurons against Bax-mediated apoptosis. J Biol Chem. 2001;276:39145–39149. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous