

Circuitry and dynamics of human transcription factor regulatory networks
- PMID: 22959076
- PMCID: PMC3679407
- DOI: 10.1016/j.cell.2012.04.040
Circuitry and dynamics of human transcription factor regulatory networks
Abstract
The combinatorial cross-regulation of hundreds of sequence-specific transcription factors (TFs) defines a regulatory network that underlies cellular identity and function. Here we use genome-wide maps of in vivo DNaseI footprints to assemble an extensive core human regulatory network comprising connections among 475 sequence-specific TFs and to analyze the dynamics of these connections across 41 diverse cell and tissue types. We find that human TF networks are highly cell selective and are driven by cohorts of factors that include regulators with previously unrecognized roles in control of cellular identity. Moreover, we identify many widely expressed factors that impact transcriptional regulatory networks in a cell-selective manner. Strikingly, in spite of their inherent diversity, all cell-type regulatory networks independently converge on a common architecture that closely resembles the topology of living neuronal networks. Together, our results provide an extensive description of the circuitry, dynamics, and organizing principles of the human TF regulatory network.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures



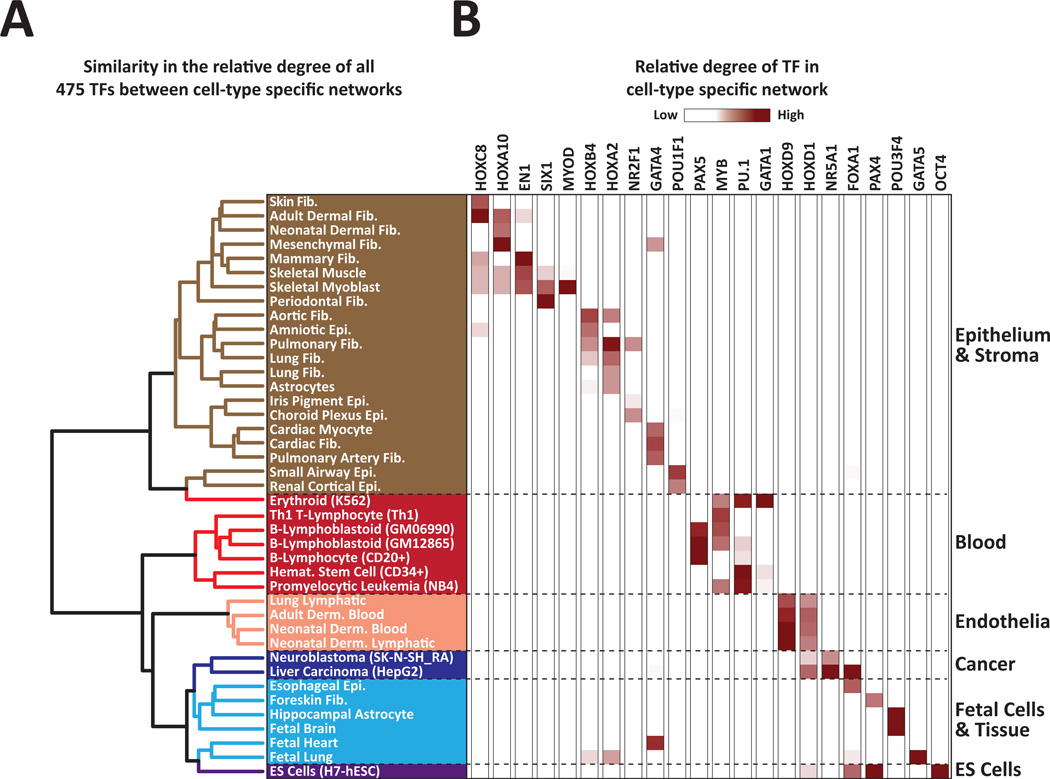
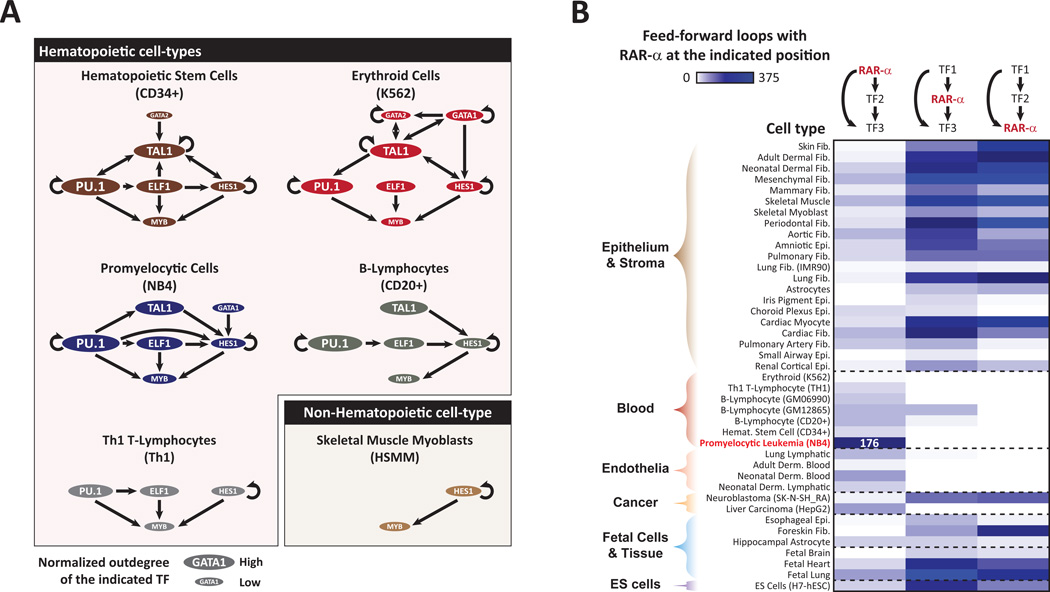
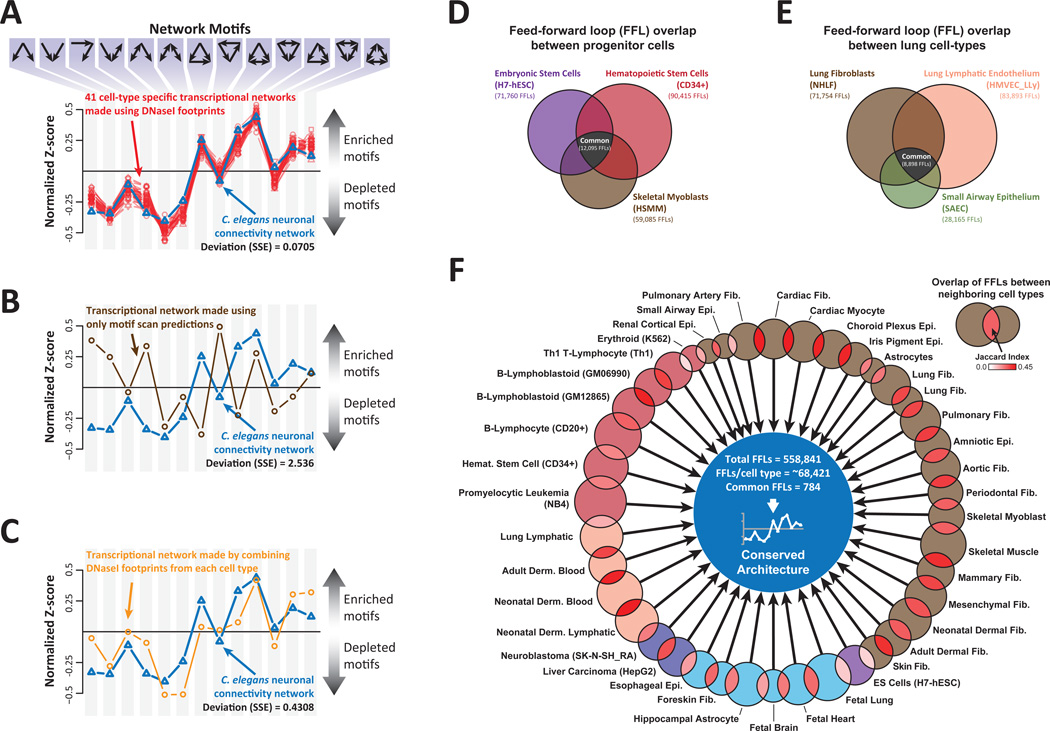
References
-
- Alon U. An Introduction to Systems Biology: Design Principles of Biological Circuits. 1 st ed. Chapman and Hall/CRC; 2006.
-
- Barabási A-L, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nature Reviews Genetics. 2004;5:101–113. - PubMed
-
- Basso K, Margolin AA, Stolovitzky G, Klein U, Dalla-Favera R, Califano A. Reverse engineering of regulatory networks in human B cells. Nature Genetics. 2005;37:382–390. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
