

A single-sample microarray normalization method to facilitate personalized-medicine workflows
- PMID: 22959562
- PMCID: PMC3508193
- DOI: 10.1016/j.ygeno.2012.08.003
A single-sample microarray normalization method to facilitate personalized-medicine workflows
Abstract
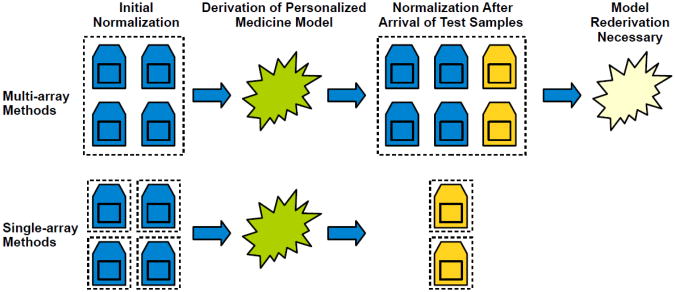
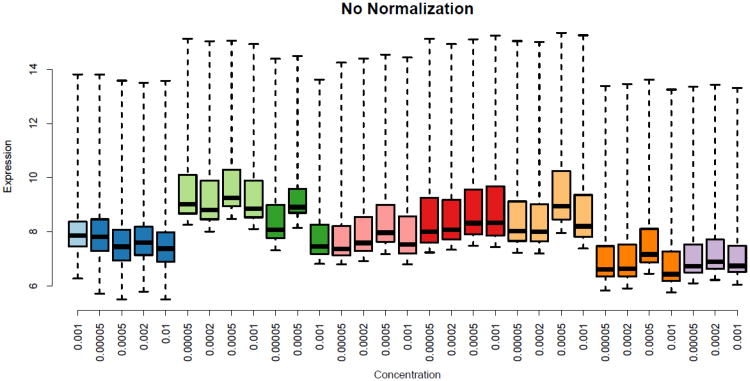
Gene-expression microarrays allow researchers to characterize biological phenomena in a high-throughput fashion but are subject to technological biases and inevitable variabilities that arise during sample collection and processing. Normalization techniques aim to correct such biases. Most existing methods require multiple samples to be processed in aggregate; consequently, each sample's output is influenced by other samples processed jointly. However, in personalized-medicine workflows, samples may arrive serially, so renormalizing all samples upon each new arrival would be impractical. We have developed Single Channel Array Normalization (SCAN), a single-sample technique that models the effects of probe-nucleotide composition on fluorescence intensity and corrects for such effects, dramatically increasing the signal-to-noise ratio within individual samples while decreasing variation across samples. In various benchmark comparisons, we show that SCAN performs as well as or better than competing methods yet has no dependence on external reference samples and can be applied to any single-channel microarray platform.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures









References
-
- van 't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AAM, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–6. - PubMed
-
- Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, Baehner FL, Walker MG, Watson D, Park T, Hiller W, Fisher ER, Wickerham DL, Bryant J, Wolmark N. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351:2817–26. - PubMed
-
- McDermott U, Downing JR, Stratton MR. Genomics and the continuum of cancer care. N Engl J Med. 2011;364:340–50. - PubMed
-
- Lo SS, Mumby PB, Norton J, Rychlik K, Smerage J, Kash J, Chew HK, Gaynor ER, Hayes DF, Epstein A, Albain KS. Prospective multicenter study of the impact of the 21-gene recurrence score assay on medical oncologist and patient adjuvant breast cancer treatment selection. J Clin Oncol. 2010;28:1671–6. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
