

Ink4-Arf locus in cancer and aging
- PMID: 22960768
- PMCID: PMC3434949
- DOI: 10.1002/wdev.40
Ink4-Arf locus in cancer and aging
Abstract
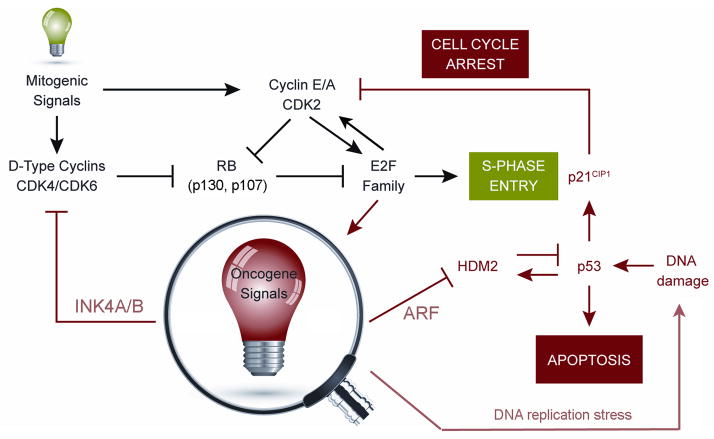
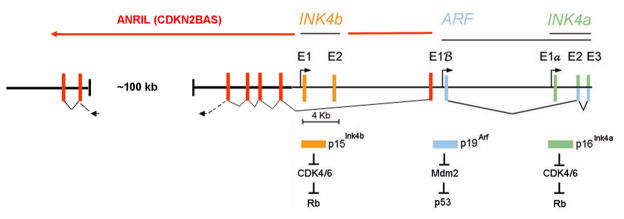
Three tumor suppressor genes at the small (<50 kb) INK4-ARF (CDKN2A/B) locus on human chromosome 9p21 coordinate a signaling network that depends on the activities of the retinoblastoma (RB) protein and the p53 transcription factor. Disruption of this circuitry, frequently by codeletion of INK4-ARF, is a hallmark of cancer, begging the question of why the intimate genetic linkage of these tumor suppressor genes has been maintained in mammals despite the risk of their coinactivation. The INK4-ARF locus is not highly expressed under normal physiologic conditions in young mammals, but its induction becomes more pronounced as animals age. Notably, INK4-ARF is actively silenced en bloc in embryonic, fetal, and adult stem cells but becomes poised to respond to oncogenic stress signals as stem cells lose their self-renewal capacity and differentiate, thereby providing a potent barrier to tumor formation. Epigenetic remodeling of the locus as a whole provides a mechanism for coordinating the activities of RB and p53. A hypothesis is that the INK4-ARF locus may have evolved to physiologically restrict the self-renewal capacities and numbers of stem and progenitor cells with the attendant consequence of limiting tissue regenerative capacity, particularly as animals age. Deletion of INK4-ARF contributes to the aberrant self-renewal capacity of tumor cells and occurs frequently in many forms of human cancer.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
