

Pathophysiology of protein aggregation and extended phenotyping in filaminopathy
- PMID: 22961544
- PMCID: PMC3437028
- DOI: 10.1093/brain/aws200
Pathophysiology of protein aggregation and extended phenotyping in filaminopathy
Abstract
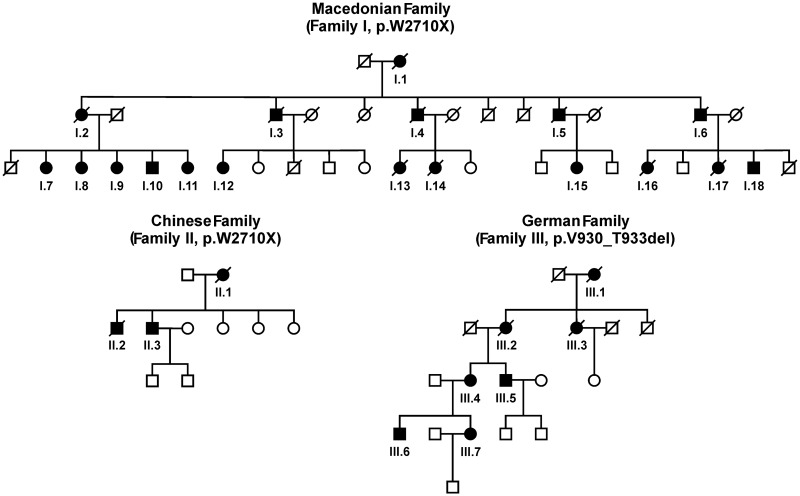
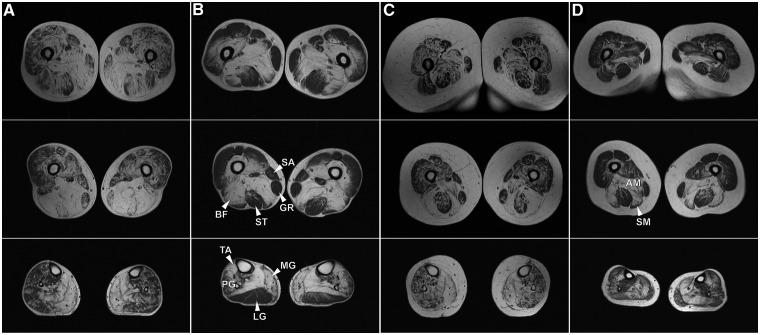
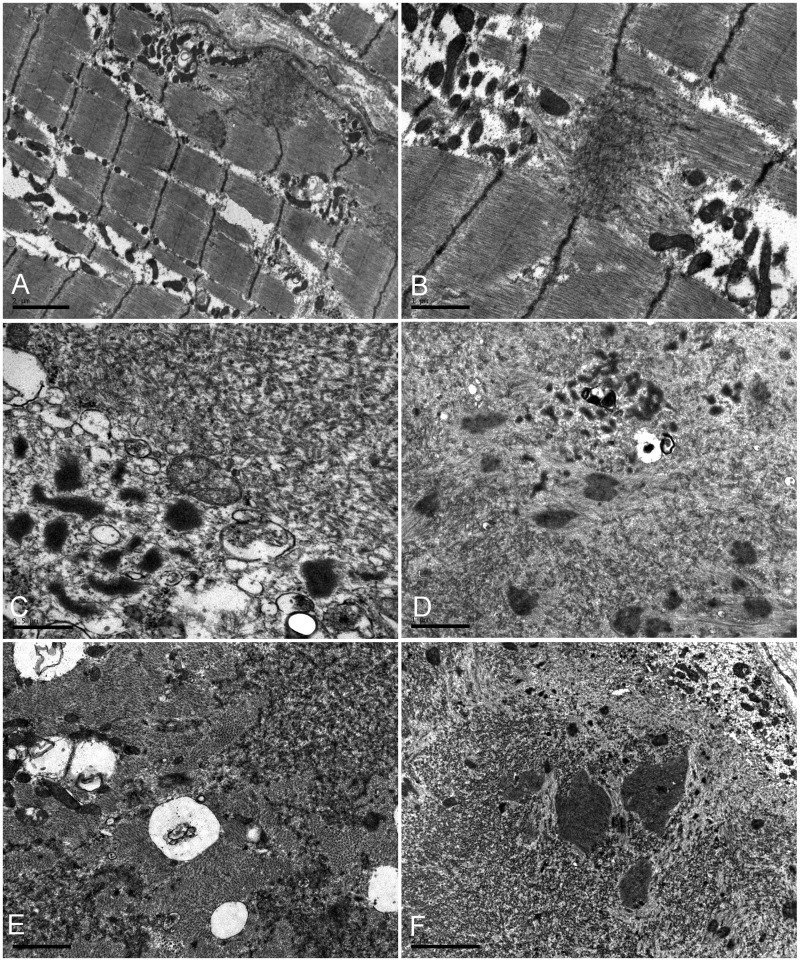
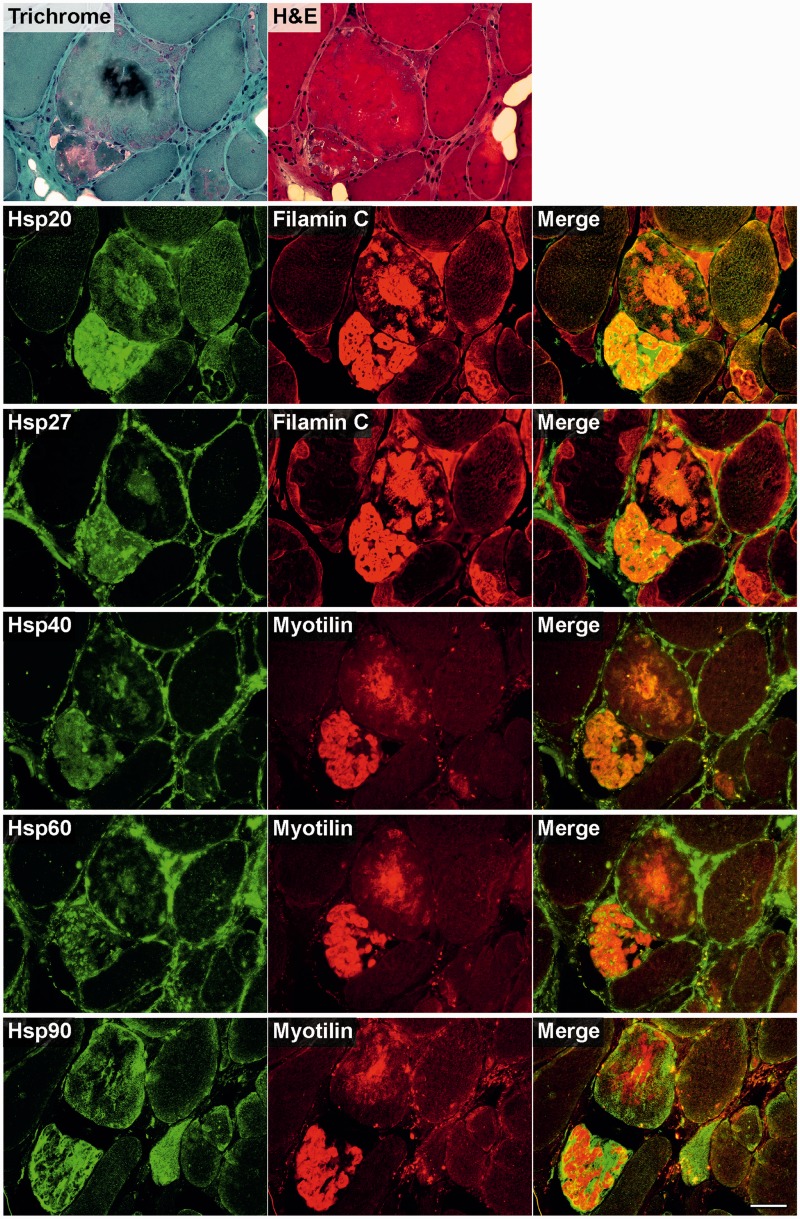
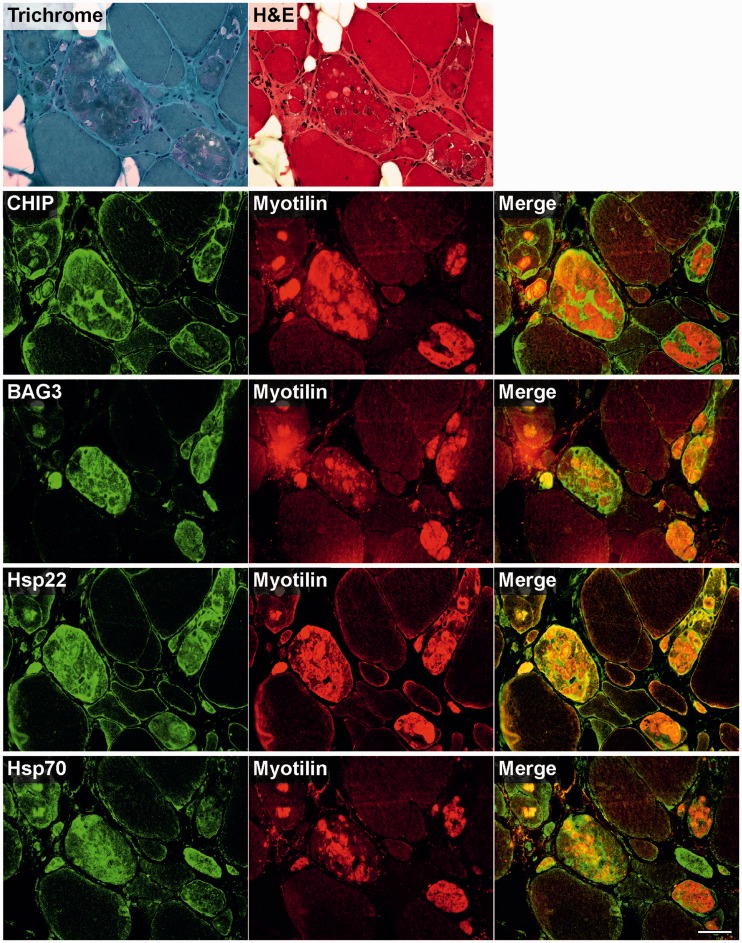
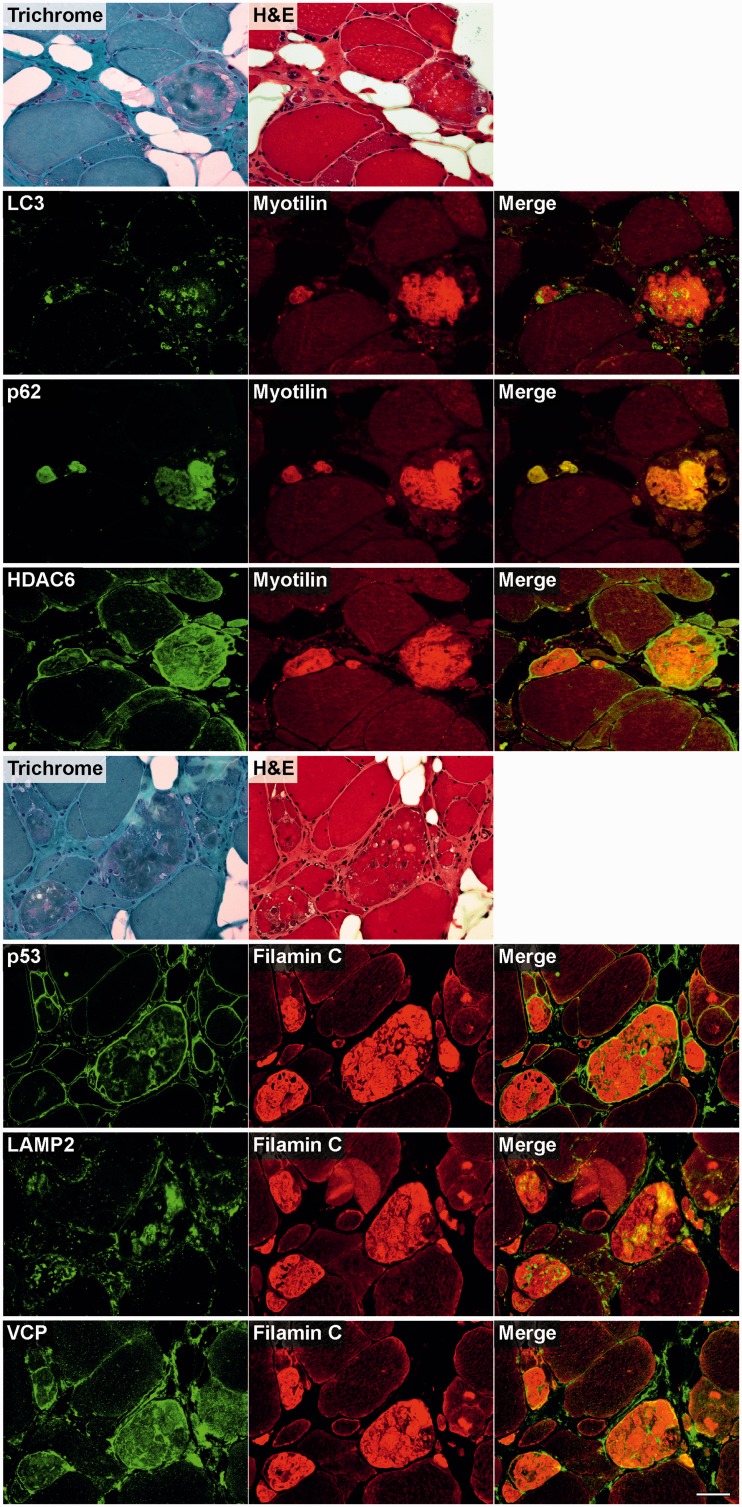
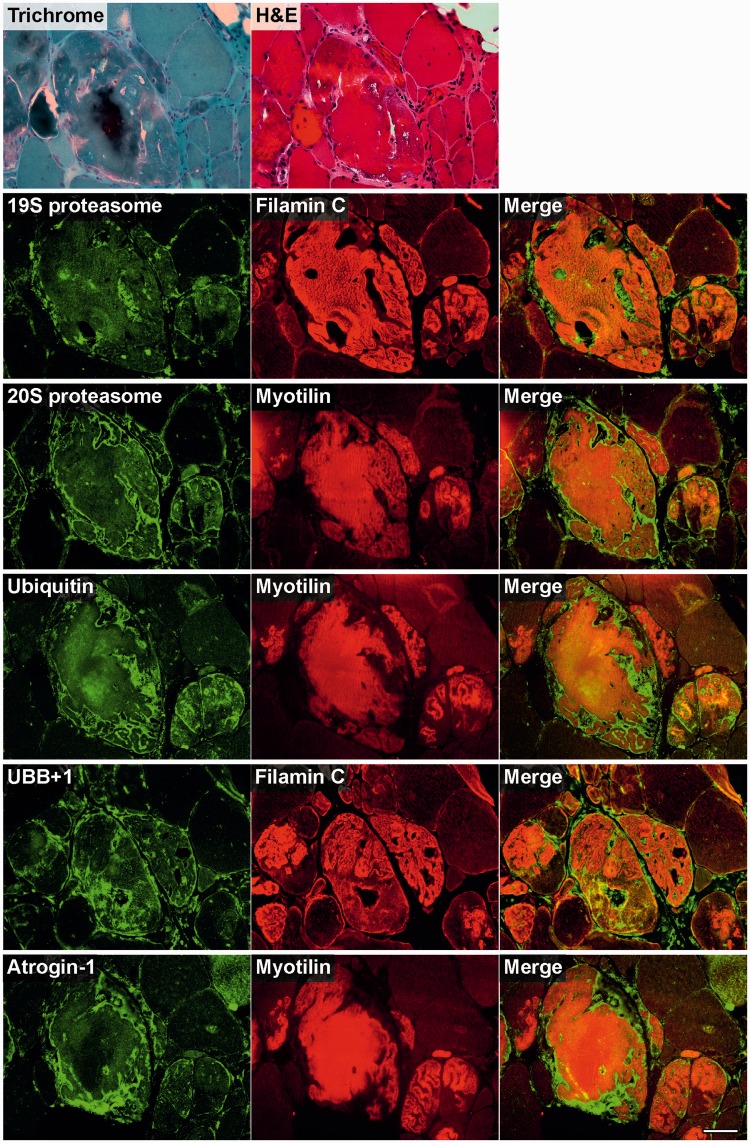
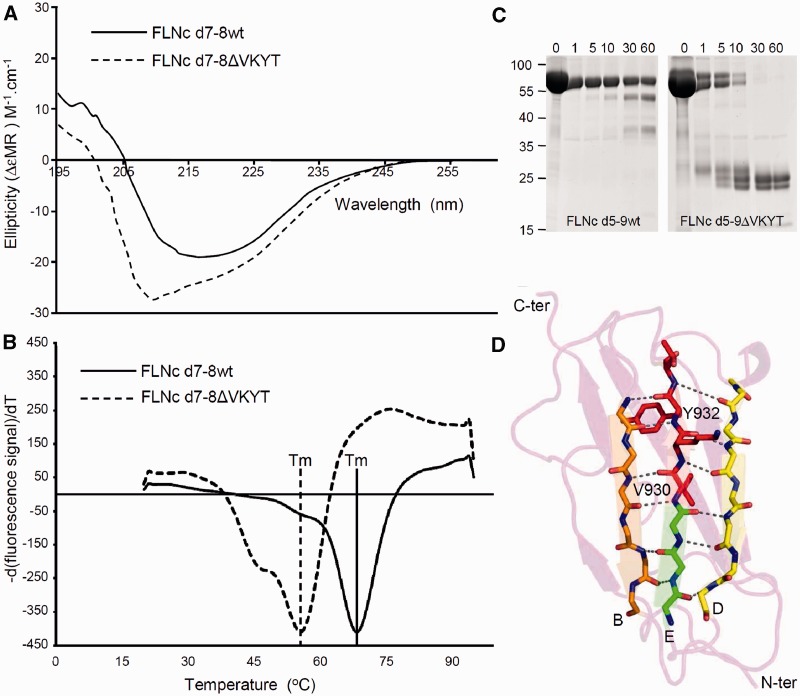
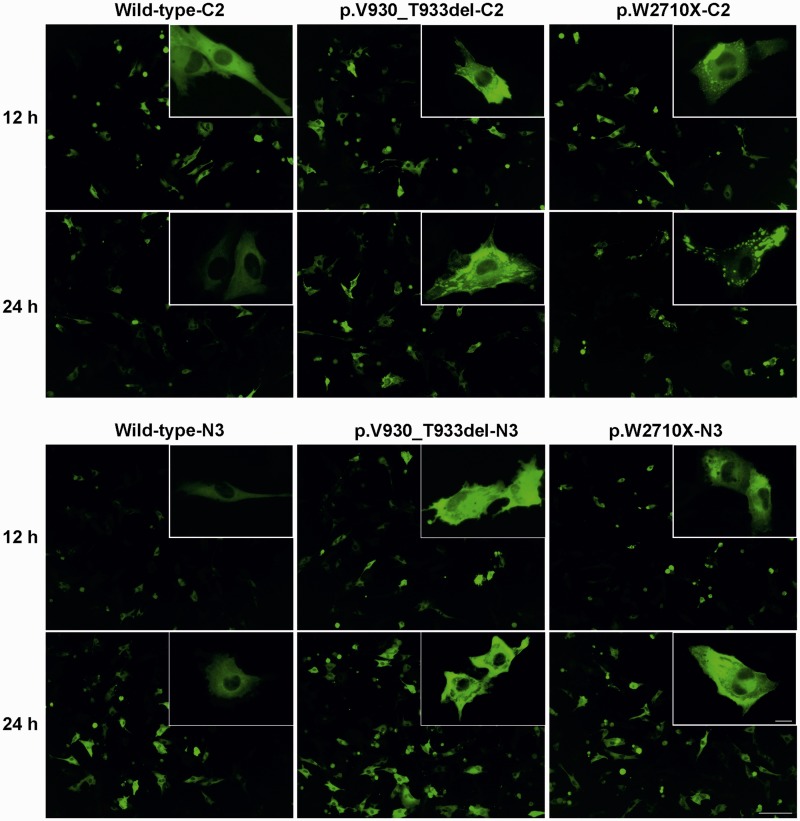
Mutations in FLNC cause two distinct types of myopathy. Disease associated with mutations in filamin C rod domain leading to expression of a toxic protein presents with progressive proximal muscle weakness and shows focal destructive lesions of polymorphous aggregates containing desmin, myotilin and other proteins in the affected myofibres; these features correspond to the profile of myofibrillar myopathy. The second variant associated with mutations in the actin-binding domain of filamin C is characterized by weakness of distal muscles and morphologically by non-specific myopathic features. A frameshift mutation in the filamin C rod domain causing haploinsufficiency was also found responsible for distal myopathy with some myofibrillar changes but no protein aggregation typical of myofibrillar myopathies. Controversial data accumulating in the literature require re-evaluation and comparative analysis of phenotypes associated with the position of the FLNC mutation and investigation of the underlying disease mechanisms. This is relevant and necessary for the refinement of diagnostic criteria and developing therapeutic approaches. We identified a p.W2710X mutation in families originating from ethnically diverse populations and re-evaluated a family with a p.V930_T933del mutation. Analysis of the expanded database allows us to refine clinical and myopathological characteristics of myofibrillar myopathy caused by mutations in the rod domain of filamin C. Biophysical and biochemical studies indicate that certain pathogenic mutations in FLNC cause protein misfolding, which triggers aggregation of the mutant filamin C protein and subsequently involves several other proteins. Immunofluorescence analyses using markers for the ubiquitin-proteasome system and autophagy reveal that the affected muscle fibres react to protein aggregate formation with a highly increased expression of chaperones and proteins involved in proteasomal protein degradation and autophagy. However, there is a noticeably diminished efficiency of both the ubiquitin-proteasome system and autophagy that impairs the muscle capacity to prevent the formation or mediate the degradation of aggregates. Transfection studies of cultured muscle cells imitate events observed in the patient's affected muscle and therefore provide a helpful model for testing future therapeutic strategies.
Figures
References
-
- Arndt V, Dick N, Tawo R, Dreiseidler M, Wenzel D, Hesse M, et al. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr Biol. 2010;20:143–8. - PubMed
-
- Bukau B, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell. 1998;92:351–66. - PubMed
-
- Carra S, Seguin SJ, Lambert H, Landry J. HspB8 chaperone activity toward poly(Q)-containing proteins depends on its association with Bag3, a stimulator of macroautophagy. J Biol Chem. 2008;283:1437–44. - PubMed
-
- Chávez Zobel AT, Loranger A, Marceau N, Thériault JR, Lambert H, Landry J. Distinct chaperone mechanisms can delay the formation of aggresomes by the myopathy-causing R120G alphaB-crystallin mutant. Hum Mol Genet. 2003;12:1609–20. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
