

Early-onset Lafora body disease
- PMID: 22961547
- PMCID: PMC3437029
- DOI: 10.1093/brain/aws205
Early-onset Lafora body disease
Abstract
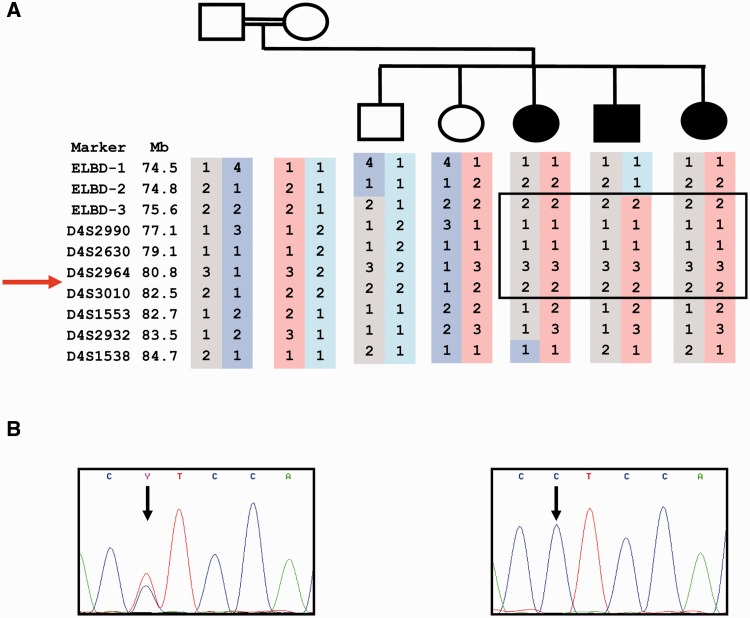
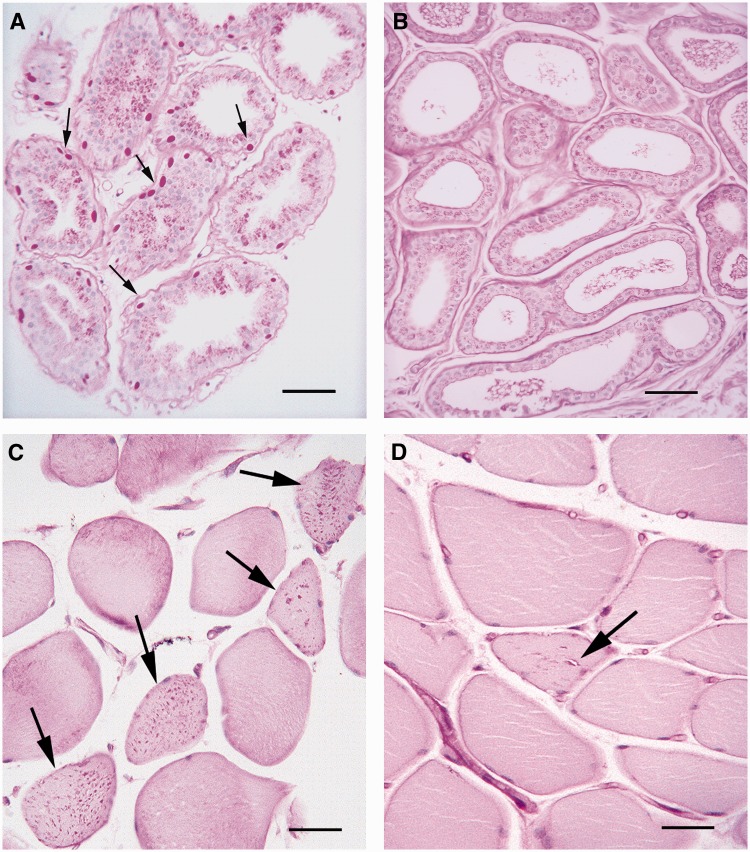
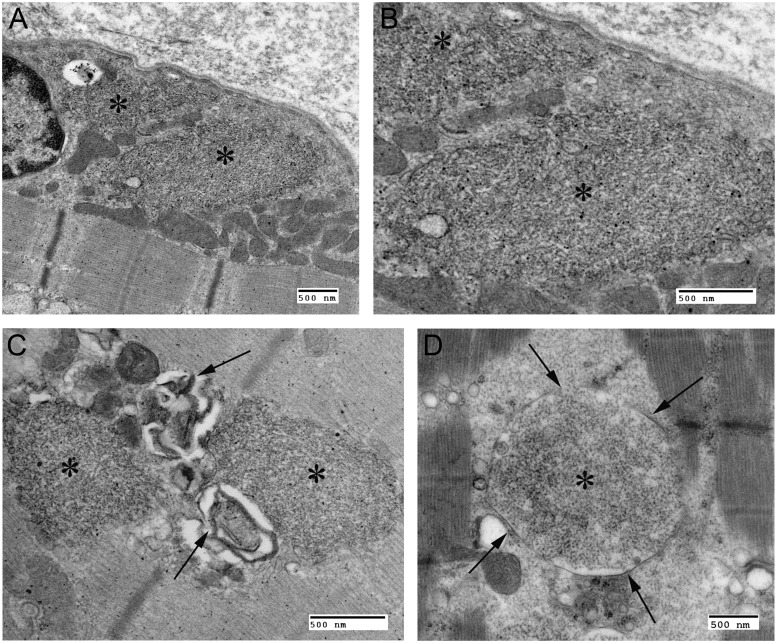
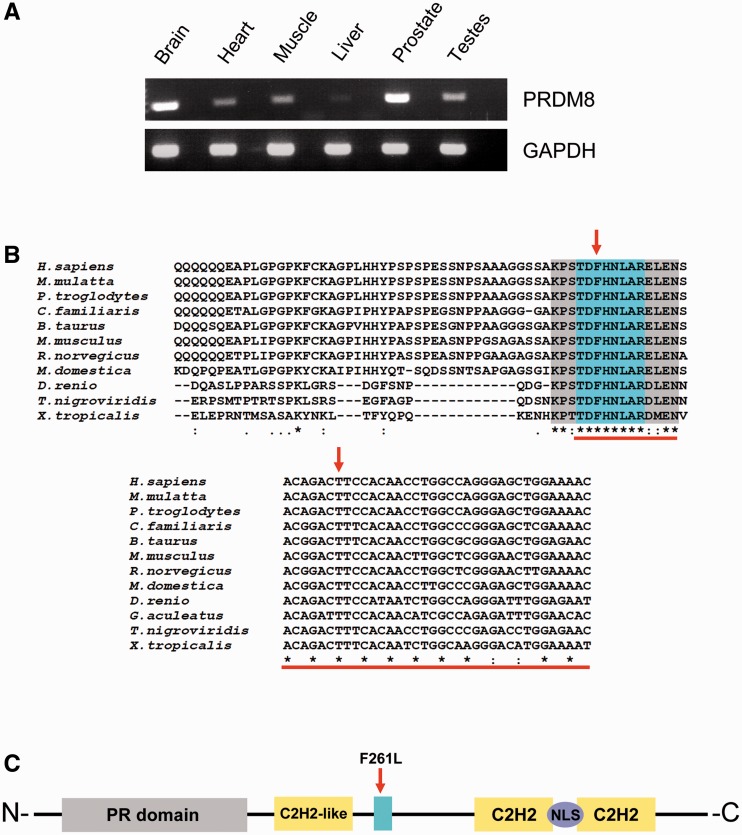
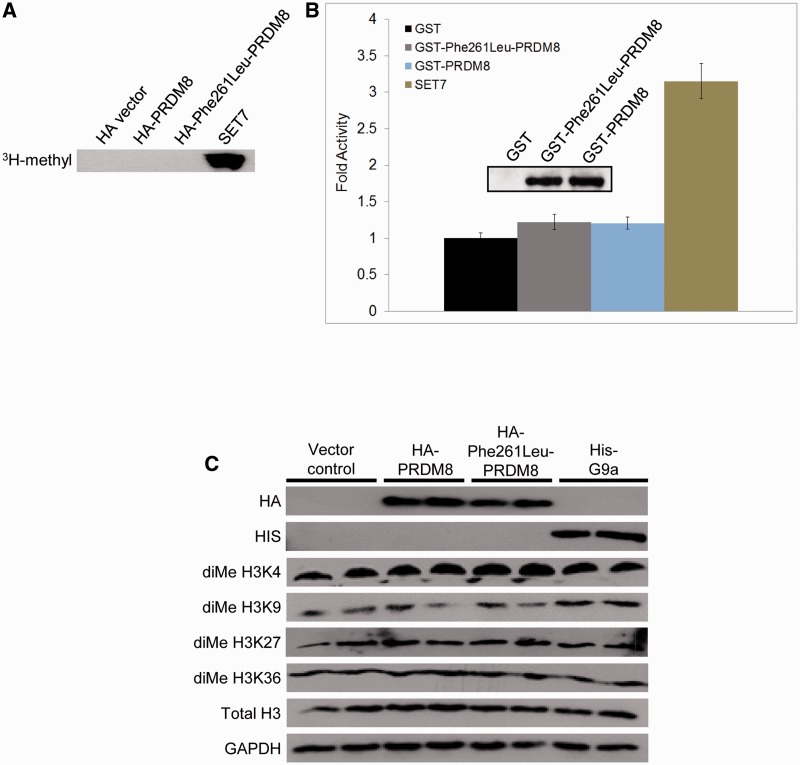
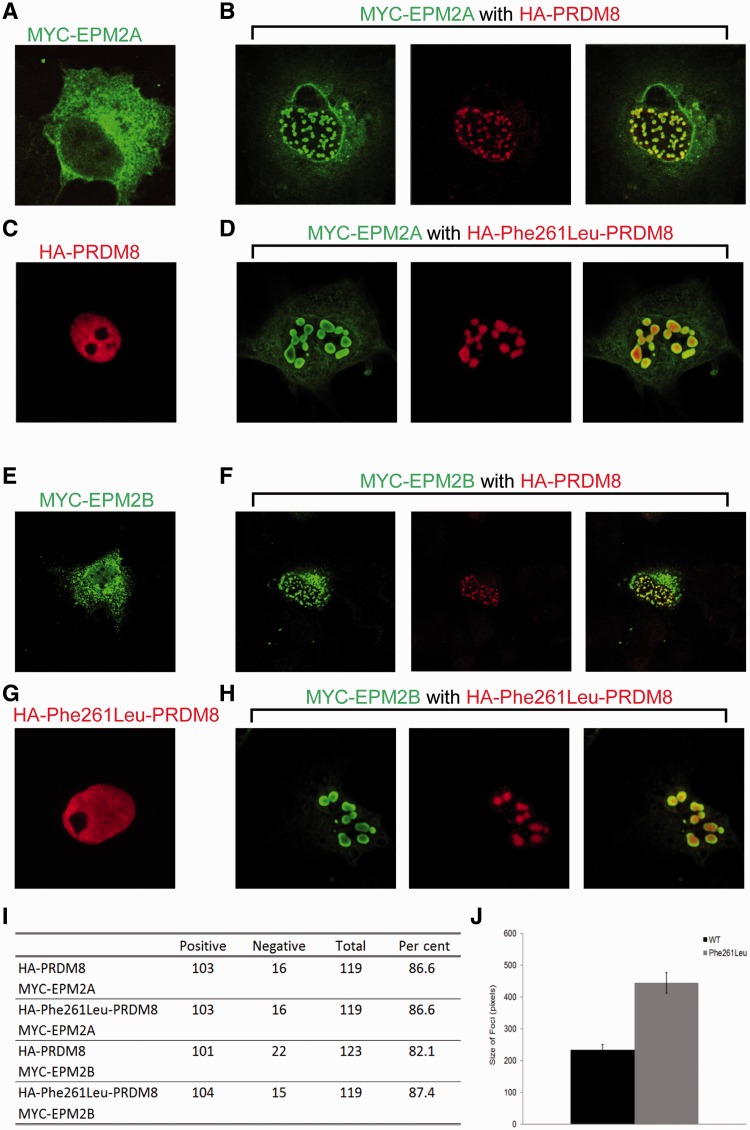
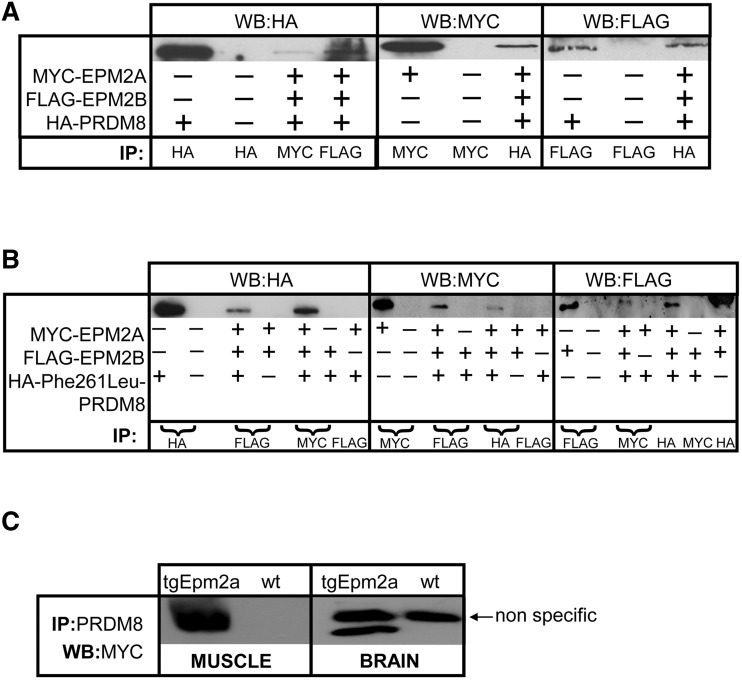
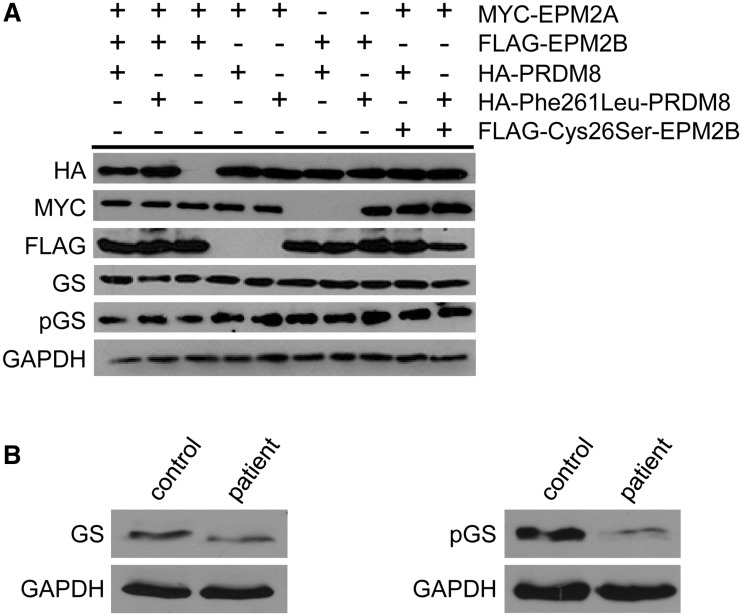
The most common progressive myoclonus epilepsies are the late infantile and late infantile-variant neuronal ceroid lipofuscinoses (onset before the age of 6 years), Unverricht-Lundborg disease (onset after the age of 6 years) and Lafora disease. Lafora disease is a distinct disorder with uniform course: onset in teenage years, followed by progressively worsening myoclonus, seizures, visual hallucinations and cognitive decline, leading to a vegetative state in status myoclonicus and death within 10 years. Biopsy reveals Lafora bodies, which are pathognomonic and not seen with any other progressive myoclonus epilepsies. Lafora bodies are aggregates of polyglucosans, poorly constructed glycogen molecules with inordinately long strands that render them insoluble. Lafora disease is caused by mutations in the EPM2A or EPM2B genes, encoding the laforin phosphatase and the malin ubiquitin ligase, respectively, two cytoplasmically active enzymes that regulate glycogen construction, ensuring symmetric expansion into a spherical shape, essential to its solubility. In this work, we report a new progressive myoclonus epilepsy associated with Lafora bodies, early-onset Lafora body disease, map its locus to chromosome 4q21.21, identify its gene and mutation and characterize the relationship of its gene product with laforin and malin. Early-onset Lafora body disease presents early, at 5 years, with dysarthria, myoclonus and ataxia. The combination of early-onset and early dysarthria strongly suggests late infantile-variant neuronal ceroid lipofuscinosis, not Lafora disease. Pathology reveals no ceroid lipofuscinosis, but Lafora bodies. The subsequent course is a typical progressive myoclonus epilepsy, though much more protracted than any infantile neuronal ceroid lipofuscinosis, or Lafora disease, patients living into the fourth decade. The mutation, c.781T>C (Phe261Leu), is in a gene of unknown function, PRDM8. We show that the PRDM8 protein interacts with laforin and malin and causes translocation of the two proteins to the nucleus. We find that Phe261Leu-PRDM8 results in excessive sequestration of laforin and malin in the nucleus and that it therefore likely represents a gain-of-function mutation that leads to an effective deficiency of cytoplasmic laforin and malin. We have identified a new progressive myoclonus epilepsy with Lafora bodies, early-onset Lafora body disease, 101 years after Lafora disease was first described. The results to date suggest that PRDM8, the early-onset Lafora body disease protein, regulates the cytoplasmic quantities of the Lafora disease enzymes.
Figures
References
-
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. - PubMed
-
- Andrade DM, Ackerley CA, Minett TS, Teive HA, Bohlega S, Scherer SW, et al. Skin biopsy in Lafora disease: genotype-phenotype correlations and diagnostic pitfalls. Neurology. 2003;61:1611–4. - PubMed
-
- Andrade DM, del Campo JM, Moro E, Minassian BA, Wennberg RA. Nonepileptic visual hallucinations in Lafora disease. Neurology. 2005;64:1311–2. - PubMed
-
- Badhwar A, Berkovic SF, Dowling JP, Gonzales M, Narayanan S, Brodtmann A, et al. Action myoclonus-renal failure syndrome: characterization of a unique cerebro-renal disorder. Brain. 2004;127:2173–82. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
